
Společnost Takeda je tradičním partnerem pacientů s hemofilií i zdravotníků, kteří o ně pečují. V České republice mohou hemofilici i lékaři profitovat z našich:
- dlouhodobých zkušeností,
- intenzivního výzkumu,
- moderních léčivých přípravků a technologií, které přinášíme na trh.
Zaměřujeme se také na optimalizaci léčby, zvyšování komfortu jejího podávání i spolupráci s odbornými společnostmi a pacientskými organizacemi. Jsme připraveni reagovat na výzvy, které léčba hematologických onemocnění přináší. V této oblasti disponujeme rozsáhlým portfoliem léčivých přípravků, zahrnujícím plazmatické, rekombinantní produkty i léčiva s prolongovaným působením.
Přinášíme sofistikovaná léčiva i pomůcky pro léčbu hemofilie všech typů a věříme, že se nám i s Vaší pomocí podaří naplnit naši vizi Bleed-Free World a že se „svět bez krvácení“ v budoucnu skutečně stane realitou. Vyzkoušejte se svými pacienty první spojení webové a mobilní aplikace v léčbě hemofilie a jediný schválený zdravotnický prostředek k individuálnímu nastavení optimální profylaxe hemofilika – webovou a mobilní aplikaci myPKFiT.
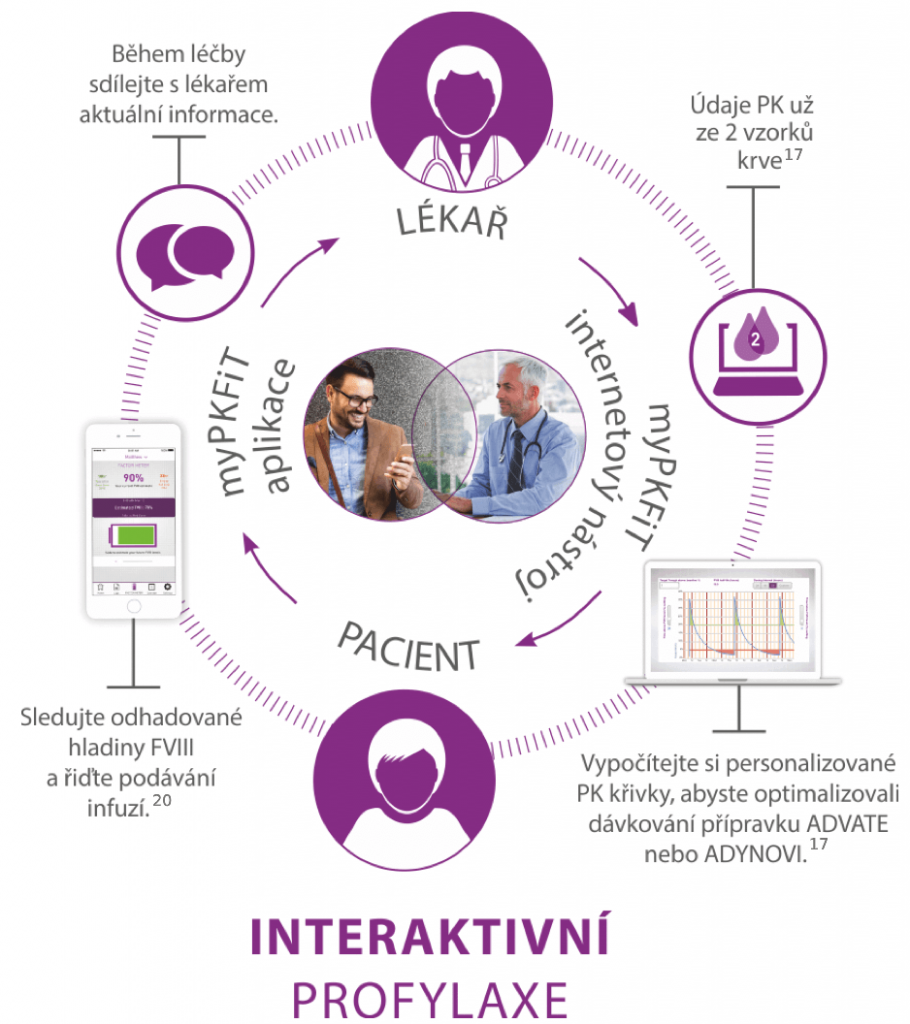
Používání aplikace myPKFiT umožňuje sdílet všechny aktualizace s ošetřujícím lékařem, optimalizovat dávkování na základě osobního farmakokinetického profilu a zároveň zvyšovat osobní účast na léčbě a samostatnost v jejím managementu. I v ČR se toto řešení setkalo s velmi příznivým ohlasem u odborníků i pacientů a velké množství z nich již výhod této technologie naplno využívá. Buďte dalšími z nich.
Stručně o hemofilii
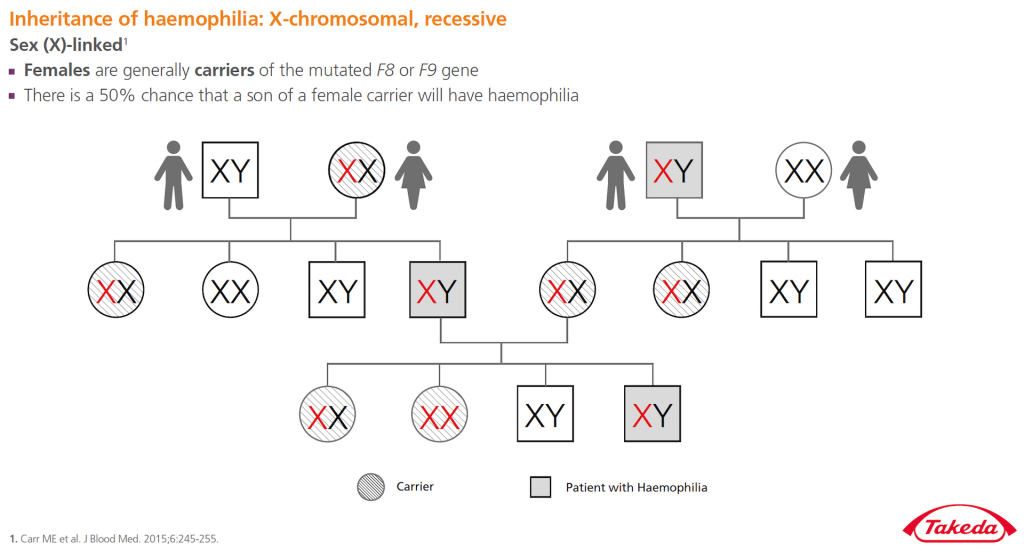
Hemofilie je vzácná, obvykle dědičná koagulopatie. Jedná se o gonosomálně recesivní onemocnění. Defektní gen je lokalizován na pohlavním chromosomu X, proto se hemofilie vyskytuje prakticky jen u mužů, u žen bývá extrémně vzácná. Hemofilie se projevuje poruchou srážlivosti krve a z toho plynoucím krvácením. Zdaleka nejčastějším typem je hemofilie typu A (deficit faktoru VIII). Druhým nejčastějším typem je typ B (deficit faktoru IX). Existují i jiná krvácivá onemocnění s deficity jiných srážecích faktorů, která se obvykle již jako hemofilie neoznačují.
Hemofilii lze odhalit již u batolat, kdy dítě začíná chodit a také padat. Ke krvácení dochází i při minimálním nárazu nebo zcela bez příčiny. Na exponovaných místech (hlava, hýždě, končetiny) se pak objevují typické podkožní a někdy i svalové hematomy. Po pádu na obličej nezřídka dochází i ke krvácení v dutině ústní. Tyto projevy může okolí rodiny mylně vyhodnotit jako týrání dítěte.
V novorozeneckém věku je krvácení poměrně vzácné. U mladších dětí a školáků může frekvenci krvácení zesílit i stres, například v období zkoušek. Pozoruje se i sezónní vliv na krvácení, například na jaře a na podzim.
Hemofilii rozdělujeme v závislosti na množství chybějícího koagulačního faktoru na:
– Těžkou formu (< 1 IU/dl nebo < 1 % normální koncentrace srážecího faktoru), při které dochází k častým spontánním krvácením především do velkých kloubů nebo svalů.1
– Středně těžkou formu (1–5 % normální koncentrace srážecího faktoru). Krvácení vzniká již po nepatrných úrazech nebo poraněních, může vzniknout i spontánně.
– Lehkou formu (> 5–40 % normální koncentrace srážecího faktoru). Ke dlouhotrvajícímu krvácení dochází po stomatologických a operačních výkonech či úrazech, ale pacient nekrvácí spontánně do kloubů nebo dalších částí těla.1
C-APROM/CZ//0701
Literatura
- Srivastava A, Brewer AK, Mauser-Bunschoten EP, et al.; Treatment Guidelines Working Group on Behalf of The World Federation Of Hemophilia. Guidelines for the management of hemophilia. Haemophilia 2013; 19 (1): e1–e47.
- Blanchette VS, Key NS, Ljung LR, et al.; Subcommittee on Factor VIII, Factor IX and Rare Coagulation Disorders of the Scientific and Standardization Committee of the International Society on Thrombosis and Hemostasis. Definitions in hemophilia: communication from the SSC of the ISTH. J Thromb Haemost 2014; 12 (11): 1935–1939.
- Rodriguez-Merchan EC. Musculoskeletal complications of hemophilia. HSS J 2010; 6 (1): 37–42.
- Aledort LM, Haschmeyer RH, Pettersson H. A longitudinal study of orthopaedic outcomes for severe factor-VIII deficient haemophiliacs. The Orthopaedic Outcome Study Group. J Intern Med 1994; 236 (4): 391–399.
- National Hemophilia Foundation. MASAC recommendation concerning prophylaxis (regular administration of clotting factor concentrate to prevent bleeding). MASAC Document #241. February 28, 2016.
- Oldenburg J, Zimmermann R, Katsarou O, et al. Controlled, cross-sectional MRI evaluation of joint status in severe haemophilia A patients treated with prophylaxis vs. on demand. Haemophilia 2015; 21 (2): 171–179.
- Pasca S, Milan M, Sarolo L, Zanon E. PK-driven prophylaxis versus standard prophylaxis: When a tailored treatment may be a real and achievable cost-saving approach in children with severe hemophilia A. Thromb Res 2017; 157: 58–63.
- Údaje z prospektivní klinické studie fáze IV s Advate (n = 66).
- Iorio A, Marcucci M, Cheng J, et al. Patient data meta-analysis of Post-Authorization Safety Surveillance (PASS) studies of haemophilia A patients treated with rAHF-PFM. Haemophilia 2014; 20 (6): 777–783.
- Pět poregistračních studií (PASS) s Advate.
- Mahdi AJ, Obaji SG, Collins PW. Role of enhanced halife factor VIII and IX in the treatment of haemophilia. Br J Haematol 2015; 169 (6): 768–776.
- Konkle BA, Stasyshyn O, Chowdary P, et al. Pegylated, full-length, recombinant factor VIII for prophylactic and on-demand treatment of severe hemophilia A. Blood 2015; 126 (9): 1078–1085.
- Valentino LA. Considerations in individualizing prophylaxis in patients with haemophilia A. Haemophilia 2014; 20 (5): 607–615.
- Windyga J, Zbikowski P, Ambroziak P, et al. Management of factor VII-deficient patients undergoing joint surgeries–preliminary results of locally developed treatment regimen. Haemophilia 2013; 19 (1): 89–93.
- Urasinski T, Stasyshyn O, Andreeva T, et al. Recombinant factor IX (BAX326) in previously treated paediatric patients with haemophilia B: a prospective clinical trial. Haemophilia 2015; 21 (2): 196–203.
- Gouw SC, van der Bom JG, van den Berget HM. Treatment-related risk factors of inhibitor development in previously untreated patients with hemophilia A: the CANAL cohort study. Blood 2007; 109 (11): 4648–4654.
- Coppola A, Di Capua M, Di Minno MN, et al. Treatment of hemophilia: a review of current advances and ongoing issues. J Blood Med 2010; 1: 183–195.
- Morfini M, Auerswald G, Kobelt RA, et al. Prophylactic treatment of haemophilia patients with inhibitors: clinical experience with recombinant factor VIIa in European Haemophilia Centres. Haemophilia 2007; 13 (5): 502–507.
- Doc. MUDr. Zuzana Čermáková, Ph.D. – Centrum pro trombózu a hemostázu, hemofilické centrum, Krevní centrum FN Ostrava, Farmakologický ústav, LF MU v Brně.
- Čermáková Z, Hrdličková R, Smejkal P, et al.; Pracovní skupina ČNHP pro standardy. Diagnostika a léčba získané hemofilie – konsenzuální doporučení Českého národního hemofilického programu (ČNHP). Transfuze Hematol dnes 2017; 23 (2): 101–109.