
I hematologové si mohou hrát…aneb reportáž ze sympozia Takeda na Pařízkách 2022
Netradiční podobu mělo sympozium, které během březnových Pařízkových dnů podpořila společnost Takeda. Jeho název byl Hematologie hrou aneb 24 zajímavých otázek a odpovědí. Organizátorům se podařilo spojit atraktivní formát připomínající televizní vědomostní soutěž s odborně přínosným obsahem. „Neměla by to být to žádná laciná show, jde o seriózní otázky se seriózními odpověďmi,“ uvedl moderátor této části programu doc. MUDr. Petr Dulíček, Ph.D.

Foto: Hana Chybová
Sympozium bylo koncipováno jako soutěž dvou týmů. Tým žlutý nastoupil ve složení MUDr. Eva Drbohlavová, RNDr. Ingrid Hrachovinová, Ph.D., MUDr. Radomíra Hrdličková a MUDr. Ester Zápotocká. Zelený tým pak tvořili MUDr. Jaromír Gumulec, Ph.D., MUDr. Zdenka Hajšmanová, MUDr. Světlana Köhlerová a MUDr. Petr Smejkal, Ph.D.
Každý tým si vybíral postupně z otázek s různou bodovou dotací z některého ze šesti okruhů: hemofilie obecně, získaná hemofilie, laboratorní diagnostika, urgentní situace, mezioborová problematika a k určitému odlehčení pak sloužily otázky mimo obor. Soutěžící si vždy mohli zvolit jednu ze čtyř nabízených možností. Při správné odpovědi se body přičítaly, při špatné odečítaly.
Úlohy moderátora se zcela profesionálně ujal doc. MUDr. Petr Dulíček, Ph.D. Spolu s tím zvládl vykonávat i funkci rozhodčího a námitky řešil rychle a nekompromisně. U čistě odborných otázek takové razance bylo zapotřebí jen výjimečně, zde se obvykle soutěžící snadno dohodli a případná zaváhání se týkala jen detailů. Například u nemocného s těžkou formou hemofilie A, který podstupuje náhradu kyčelního kloubu, se jednoznačně shodli na nutnosti profylaxe. Lišili se ale v názoru na požadovanou aktivitu FVIII při profylaxi. Zatímco žlutý tým se klonil k hodnotě 100 procent, doc. Dulíček argumentoval, proč je podle něj vhodnější hodnota 150 procent. „Hodnota 100 procent nemusí být dostatečná, je to velmi invazivní výkon. Bohužel také platí, že při náhradě kyčelního kloubu mnohokrát profylaxi dáme, ale pak s ní příliš brzy ustupujeme.“
Podobně bez kontroverzí se soutěžící sjednotili na stanovisku hematologa u gravidní přenašečky hemofilie A s hodnotou FVIII 45 procent. Zde se zelený tým shodl na tom, že se bude řídit hlavně podle hodnoty FVIII měsíc před porodem a podle ní zvolí případnou substituci. „Tím by se mělo předejít prolongovanému krvácení,“ souhlasil z pozice arbitra doc. Dulíček.
Několik otázek se týkalo postupu u nemocných na antikoagulační léčbě. Jedna z nich předkládala kazuistiku 70letého muže, který byl přivezen na urgentní příjem se zlomeninou krčku. Užívá pro fibrilaci síní rivaroxaban 20 mg jednou denně, poslední dávku užil před dvěma hodinami. Operaci nelze odkládat vzhledem k (ne)dostupnosti anesteziologie. Jak se s touto situací vyrovnat z pohledu hematologa? Zde si zelený tým správě zvolil možnosti „Zajistit PT, aPTT a krevní obraz a operaci provést bez znalosti výsledku, podat koncentrát protrombinového komplexu v dávce 50 jednotek na kilogram.“
Podobně se bez velkých diskusí žlutý tým vyrovnal s otázkou, jaký význam má pátrání po příčině získané hemofillie. Zde byla správná možnost: „Velký, protože významná část případů je sekundární při nádorovém onemocnění, tito nemocní mají vyšší riziko krvácení a horší prognózu, není-li vyřešena základní příčina.“ To doc. Dulíček komentoval: „Tak jako jinde v medicíně, i zde platí, že je dobré po příčině pátrat, protože pak je nižší riziko relapsu.“
Kamenem úrazu byly spíše otázky mimo obor. U nich se také nejvíc uplatňoval doc. Dulíček v roli rázného arbitra. Například nenechal příliš prostoru pro diskusi, jak autoři otázek přišli na to, že k vysátí veškeré krve z člověka je potřeba 1 200 000 komárů. Zelený tým ztratil body na otázce, kolik srdcí má chobotnice. Jeho tip, že jedno, se neukázal jako správný. Tento hlavonožec má dvě brachiální srdce uložená po stranách těla, ta okysličují krev tak, že ji čerpají přes cévy v žábrách. Systémové srdce ve středu těla pumpuje okysličenou krev do zbytku organismu. „To dá rozum, že tvor, který vypadá jako chobotnice, nemůže mít jedno srdce,“ glosoval doc. Dulíček.
I přes toto zaváhání se nakonec zelený tým stal celkovým vítězem – žlutý tým to bral sportovně, celou soutěž si evidentně užil také, stejně jako diváci v sále.
Redakce Medical tribune 3.2022
C-APROM/CZ/HEM/0042
Datum přípravy 4/2022
Nové výzvy v léčbě chronické hypoparatyreózy
Společnost Takeda dne 25.3.2021 připravila on-line edukační seminář NOVÉ VÝZVY V LÉČBĚ CHRONICKÉ HYPOPARATYREÓZY, jehož cílem bylo sdílení zkušeností týkající se diagnostiky a léčby chronické hypoparatyreózy.
Pozvání na seminář přijala Dr. Karin Amrein (Rakousko), která má bohaté klinické zkušenosti s léčbou rhPTH (1-84) a prof. Michal Kršek, který je jedním z autorů Doporučených postupů pro diagnostiku a léčbu chronické hypoparatyreózy.
Více informací pro Vaše pacienty www.hypopara.cz.
Mohlo by Vás zajímat




NATPAR
Zkrácené informace o léčivém přípravku
▼ Tento léčivý přípravek podléhá dalšímu sledování. To umožní rychlé získání nových informací o bezpečnosti. Žádáme zdravotnické pracovníky, aby hlásili jakákoli podezření na nežádoucí účinky. Podrobnosti o hlášení nežádoucích účinků viz bod 4.8 SPC.
Název přípravku: Natpar 25 mikrogramů/dávka prášek a rozpouštědlo pro injekční roztok; Natpar 50 mikrogramů/dávka prášek a rozpouštědlo pro injekční roztok; Natpar 75 mikrogramů/dávka prášek a rozpouštědlo pro injekční roztok; Natpar 100 mikrogramů/dávka prášek a rozpouštědlo pro injekční Složení: Jedna dávka obsahuje po rekonstituci 25/50/75/100 mikrogramů hormonum parathyroidum (rDNA)* v 71,4 mikrolitrech roztoku. Jedna zásobní vložka obsahuje 350/700/1050/1400 mikrogramů hormonum parathyroidum (rDNA). *Parathormon (rDNA) produkovaný E. coli pomocí rekombinantní DNA technologie je identický se sekvencí 84 aminokyselin endogenního lidského parathormonu. Indikace: Přípravek Natpar je indikován jako přídatná léčba dospělých pacientů s chronickou hypoparathyreózou, kteří nejsou dostatečně kontrolováni samostatnou standardní léčbou. Dávkování a způsob podání: Zahajte léčbu 50 mikrogramy jednou denně subkutánní injekcí do stehna (střídejte stehna každý den). Pokud je před podáním dávky sérové kalcium > 2,25 mmol/l, je možné zvážit podání počáteční dávky 25 mikrogramů. U pacientů užívajících aktivní vitamin D snižte jeho dávku o 50 %, pokud je sérové kalcium před podáním dávky nad 1,87 mmol/l. Udržujte doplňkovou dávku kalcia. Změřte sérovou koncentraci kalcia během 2 až 5 dnů před podáním dávky. Pokud je sérová hladina kalcia před dávkou pod 1,87 mmol/l nebo nad 2,55 mmol/l, mělo by se měření opakovat následující den. Upravte dávku aktivního vitamínu D nebo doplňku kalcia nebo obou na základě sérové hladiny kalcia a klinického hodnocení. Úpravy se mají opakovat, dokud nebudou cílové hladiny sérového kalcia před dávkou v rozsahu 2,0 - 2,25 mmol/l, aktivní vitamín D bude ukončen a doplněk kalcia je dostatečný pro splnění denních požadavků. Během titrace musí být monitorována koncentrace sérového kalcia. Dávka přípravku Natpar se může zvýšit o 25 mikrogramů přibližně každé 2 až 4 týdny, do maximální denní dávky 100 mikrogramů. K titraci dávky dolů na minimum 25 mikrogramů může dojít kdykoliv. Pokyny k úpravě dávky po úvodním období – viz SPC. Přípravek Natpar je vhodný pro podávání samotným pacientem jako subkutánní injekce jednou denně střídavě do každého stehna. Kontraindikace: Hypersenzitivita na léčivou látku nebo na kteroukoli pomocnou látku. U pacientů, kteří podstupují nebo dříve podstupovali radioterapii skeletu, s malignitami skeletu nebo kostními metastázami, kteří mají zvýšené výchozí riziko vzniku osteosarkomu (pacienti s Pagetovou chorobou kostí nebo dědičnými poruchami), s nevysvětleným zvýšením kostní alkalické fosfatázy a s pseudohypoparathyreózou. Zvláštní upozornění a opatření pro použití: Monitorování pacientů během léčby: Sérové hladiny kalcia před dávkou a v některých případech po dávce musí být během léčby přípravkem Natpar sledovány. Hyperkalcemie: často se objevovala během titračního období. V případě závažné hyperkalcemie (> 3,0 mmol/l nebo nad horní hranici normy s příznaky), zvažte hydrataci a dočasné zastavení podávání přípravku Natpar, kalcia a aktivního vitamínu D, dokud se sérové kalcium nevrátí do normálního rozsahu. Hypokalcemie: Riziko závažné hypokalcemie je nejvyšší po odložení podání, vynechání nebo náhlém vysazení přípravku Natpar, může se ale vyskytnout kdykoli. Souběžné použití se srdečními glykosidy: Hyperkalcemie z jakékoliv příčiny může predisponovat k toxicitě digitalis. Závažná porucha funkce ledvin nebo jater: Používat s opatrností u pacientů se závažnou poruchou ledvin nebo jater. Použití u mladých dospělých: Používat s opatrností u mladých dospělých pacientů s otevřenými epifýzami, protože tito pacienti mohou být vystaveni zvýšenému riziku vzniku osteosarkomu. Použití u starších pacientů: Klinické studie nezahrnovaly dostatečný počet subjektů ve věku ≥ 65 let pro stanovení, zda je odpověď u těchto subjektů odlišná od mladších subjektů. Tachyfylaxe: Účinek přípravku Natpar se může po čase u některých pacientů snížit. Urolitiáza: U pacientů s aktivní nebo nedávnou urolitiázou je nutno používat přípravek Natpar s opatrností vzhledem k jeho potenciálu tento stav zhoršovat. Hypersenzitivita: Po uvedení na trh byly hlášeny hypersenzitivní reakce, které mohou zahrnovat anafylaxi, dyspnoi, angioedém, kopřivku, vyrážku atd. Interakce: Kombinované použití přípravku Natpar a srdečních glykosidů může pacienty predisponovat k toxicitě digitalisu, pokud vznikne hyperkalcémie. Současné používání přípravku Natpar s bisfosfonáty se nedoporučuje. Fertilita, těhotenství a kojení: Údaje o podávání přípravku Natpar těhotným ženám nejsou k dispozici. Studie reprodukční toxicity na zvířatech nenaznačují přímé nebo nepřímé škodlivé účinky. Není známo, zda se přípravek Natpar vylučuje do lidského mateřského mléka. Dostupné farmakologické údaje u zvířat prokázaly vylučování přípravku Natpar do mléka. Neexistují žádné údaje u člověka o vlivu přípravku Natpar na plodnost. Studie na zvířatech nenaznačují žádnou poruchu plodnosti. Nežádoucí účinky: Nejčastější nežádoucí reakce byly hyperkalcemie, hypokalcemie a jejich související klinické projevy, zahrnující bolesti hlavy, průjem, zvracení, parestezie, hypestezie a hyperkalciurie. Ostatní nežádoucí účinky – viz SPC. Předávkování: Může způsobit hyperkalcémii. Těžká hyperkalcémie může být život ohrožující stav vyžadující neodkladnou lékařskou péči a pečlivé monitorování. Podmínky uchovávání: Uchovávejte v chladničce (2 °C – 8 °C). Chraňte před mrazem. Uchovávejte zásobní vložky v držáku v krabičce, aby byly chráněny před světlem. Podmínky uchovávání po rekonstituci – viz SPC. Držitel rozhodnutí o registraci: Shire Pharmaceuticals Ireland Limited, Block 2 & 3 Miesian Plaza, 50 – 58 Baggot Street Lower, Dublin 2, Irsko. Registrační čísla: EU/1/15/1078/001-004. Poslední revize SPC: 04/2020.
Výdej léčivého přípravku je vázán na lékařský předpis. Léčivý přípravek není hrazen z prostředků veřejného zdravotního pojištění. Před předepsáním se seznamte s úplným zněním Souhrnu údajů o přípravku.
Žádáme zdravotnické pracovníky, aby hlásili podezření na nežádoucí účinky na SÚKL nebo společnosti Takeda emailem na AE.CZE@takeda.com. Podezření na nežádoucí účinky hlaste také podle národních legislativních požadavků.
▼ Tento léčivý přípravek podléhá dalšímu sledování. To umožní rychlé získání nových informací o bezpečnosti. Žádáme zdravotnické pracovníky, aby hlásili jakákoli podezření na nežádoucí účinky. Podrobnosti o hlášení nežádoucích účinků viz bod 4.8 SPC.
Název přípravku: Natpar 25 mikrogramů/dávka prášek a rozpouštědlo pro injekční roztok; Natpar 50 mikrogramů/dávka prášek a rozpouštědlo pro injekční roztok; Natpar 75 mikrogramů/dávka prášek a rozpouštědlo pro injekční roztok; Natpar 100 mikrogramů/dávka prášek a rozpouštědlo pro injekční Složení: Jedna dávka obsahuje po rekonstituci 25/50/75/100 mikrogramů hormonum parathyroidum (rDNA)* v 71,4 mikrolitrech roztoku. Jedna zásobní vložka obsahuje 350/700/1050/1400 mikrogramů hormonum parathyroidum (rDNA). *Parathormon (rDNA) produkovaný E. coli pomocí rekombinantní DNA technologie je identický se sekvencí 84 aminokyselin endogenního lidského parathormonu. Indikace: Přípravek Natpar je indikován jako přídatná léčba dospělých pacientů s chronickou hypoparathyreózou, kteří nejsou dostatečně kontrolováni samostatnou standardní léčbou. Dávkování a způsob podání: Zahajte léčbu 50 mikrogramy jednou denně subkutánní injekcí do stehna (střídejte stehna každý den). Pokud je před podáním dávky sérové kalcium > 2,25 mmol/l, je možné zvážit podání počáteční dávky 25 mikrogramů. U pacientů užívajících aktivní vitamin D snižte jeho dávku o 50 %, pokud je sérové kalcium před podáním dávky nad 1,87 mmol/l. Udržujte doplňkovou dávku kalcia. Změřte sérovou koncentraci kalcia během 2 až 5 dnů před podáním dávky. Pokud je sérová hladina kalcia před dávkou pod 1,87 mmol/l nebo nad 2,55 mmol/l, mělo by se měření opakovat následující den. Upravte dávku aktivního vitamínu D nebo doplňku kalcia nebo obou na základě sérové hladiny kalcia a klinického hodnocení. Úpravy se mají opakovat, dokud nebudou cílové hladiny sérového kalcia před dávkou v rozsahu 2,0 - 2,25 mmol/l, aktivní vitamín D bude ukončen a doplněk kalcia je dostatečný pro splnění denních požadavků. Během titrace musí být monitorována koncentrace sérového kalcia. Dávka přípravku Natpar se může zvýšit o 25 mikrogramů přibližně každé 2 až 4 týdny, do maximální denní dávky 100 mikrogramů. K titraci dávky dolů na minimum 25 mikrogramů může dojít kdykoliv. Pokyny k úpravě dávky po úvodním období – viz SPC. Přípravek Natpar je vhodný pro podávání samotným pacientem jako subkutánní injekce jednou denně střídavě do každého stehna. Kontraindikace: Hypersenzitivita na léčivou látku nebo na kteroukoli pomocnou látku. U pacientů, kteří podstupují nebo dříve podstupovali radioterapii skeletu, s malignitami skeletu nebo kostními metastázami, kteří mají zvýšené výchozí riziko vzniku osteosarkomu (pacienti s Pagetovou chorobou kostí nebo dědičnými poruchami), s nevysvětleným zvýšením kostní alkalické fosfatázy a s pseudohypoparathyreózou. Zvláštní upozornění a opatření pro použití: Monitorování pacientů během léčby: Sérové hladiny kalcia před dávkou a v některých případech po dávce musí být během léčby přípravkem Natpar sledovány. Hyperkalcemie: často se objevovala během titračního období. V případě závažné hyperkalcemie (> 3,0 mmol/l nebo nad horní hranici normy s příznaky), zvažte hydrataci a dočasné zastavení podávání přípravku Natpar, kalcia a aktivního vitamínu D, dokud se sérové kalcium nevrátí do normálního rozsahu. Hypokalcemie: Riziko závažné hypokalcemie je nejvyšší po odložení podání, vynechání nebo náhlém vysazení přípravku Natpar, může se ale vyskytnout kdykoli. Souběžné použití se srdečními glykosidy: Hyperkalcemie z jakékoliv příčiny může predisponovat k toxicitě digitalis. Závažná porucha funkce ledvin nebo jater: Používat s opatrností u pacientů se závažnou poruchou ledvin nebo jater. Použití u mladých dospělých: Používat s opatrností u mladých dospělých pacientů s otevřenými epifýzami, protože tito pacienti mohou být vystaveni zvýšenému riziku vzniku osteosarkomu. Použití u starších pacientů: Klinické studie nezahrnovaly dostatečný počet subjektů ve věku ≥ 65 let pro stanovení, zda je odpověď u těchto subjektů odlišná od mladších subjektů. Tachyfylaxe: Účinek přípravku Natpar se může po čase u některých pacientů snížit. Urolitiáza: U pacientů s aktivní nebo nedávnou urolitiázou je nutno používat přípravek Natpar s opatrností vzhledem k jeho potenciálu tento stav zhoršovat. Hypersenzitivita: Po uvedení na trh byly hlášeny hypersenzitivní reakce, které mohou zahrnovat anafylaxi, dyspnoi, angioedém, kopřivku, vyrážku atd. Interakce: Kombinované použití přípravku Natpar a srdečních glykosidů může pacienty predisponovat k toxicitě digitalisu, pokud vznikne hyperkalcémie. Současné používání přípravku Natpar s bisfosfonáty se nedoporučuje. Fertilita, těhotenství a kojení: Údaje o podávání přípravku Natpar těhotným ženám nejsou k dispozici. Studie reprodukční toxicity na zvířatech nenaznačují přímé nebo nepřímé škodlivé účinky. Není známo, zda se přípravek Natpar vylučuje do lidského mateřského mléka. Dostupné farmakologické údaje u zvířat prokázaly vylučování přípravku Natpar do mléka. Neexistují žádné údaje u člověka o vlivu přípravku Natpar na plodnost. Studie na zvířatech nenaznačují žádnou poruchu plodnosti. Nežádoucí účinky: Nejčastější nežádoucí reakce byly hyperkalcemie, hypokalcemie a jejich související klinické projevy, zahrnující bolesti hlavy, průjem, zvracení, parestezie, hypestezie a hyperkalciurie. Ostatní nežádoucí účinky – viz SPC. Předávkování: Může způsobit hyperkalcémii. Těžká hyperkalcémie může být život ohrožující stav vyžadující neodkladnou lékařskou péči a pečlivé monitorování. Podmínky uchovávání: Uchovávejte v chladničce (2 °C – 8 °C). Chraňte před mrazem. Uchovávejte zásobní vložky v držáku v krabičce, aby byly chráněny před světlem. Podmínky uchovávání po rekonstituci – viz SPC. Držitel rozhodnutí o registraci: Shire Pharmaceuticals Ireland Limited, Block 2 & 3 Miesian Plaza, 50 – 58 Baggot Street Lower, Dublin 2, Irsko. Registrační čísla: EU/1/15/1078/001-004. Poslední revize SPC: 04/2020.
Výdej léčivého přípravku je vázán na lékařský předpis. Léčivý přípravek není hrazen z prostředků veřejného zdravotního pojištění. Před předepsáním se seznamte s úplným zněním Souhrnu údajů o přípravku.
Žádáme zdravotnické pracovníky, aby hlásili podezření na nežádoucí účinky na SÚKL nebo společnosti Takeda emailem na AE.CZE@takeda.com. Podezření na nežádoucí účinky hlaste také podle národních legislativních požadavků.
Mohlo by Vás zajímat
NATPAR
Zkrácené informace o léčivém přípravku
▼ Tento léčivý přípravek podléhá dalšímu sledování. To umožní rychlé získání nových informací o bezpečnosti. Žádáme zdravotnické pracovníky, aby hlásili jakákoli podezření na nežádoucí účinky. Podrobnosti o hlášení nežádoucích účinků viz bod 4.8 SPC.
Název přípravku: Natpar 25 mikrogramů/dávka prášek a rozpouštědlo pro injekční roztok; Natpar 50 mikrogramů/dávka prášek a rozpouštědlo pro injekční roztok; Natpar 75 mikrogramů/dávka prášek a rozpouštědlo pro injekční roztok; Natpar 100 mikrogramů/dávka prášek a rozpouštědlo pro injekční Složení: Jedna dávka obsahuje po rekonstituci 25/50/75/100 mikrogramů hormonum parathyroidum (rDNA)* v 71,4 mikrolitrech roztoku. Jedna zásobní vložka obsahuje 350/700/1050/1400 mikrogramů hormonum parathyroidum (rDNA). *Parathormon (rDNA) produkovaný E. coli pomocí rekombinantní DNA technologie je identický se sekvencí 84 aminokyselin endogenního lidského parathormonu. Indikace: Přípravek Natpar je indikován jako přídatná léčba dospělých pacientů s chronickou hypoparathyreózou, kteří nejsou dostatečně kontrolováni samostatnou standardní léčbou. Dávkování a způsob podání: Zahajte léčbu 50 mikrogramy jednou denně subkutánní injekcí do stehna (střídejte stehna každý den). Pokud je před podáním dávky sérové kalcium > 2,25 mmol/l, je možné zvážit podání počáteční dávky 25 mikrogramů. U pacientů užívajících aktivní vitamin D snižte jeho dávku o 50 %, pokud je sérové kalcium před podáním dávky nad 1,87 mmol/l. Udržujte doplňkovou dávku kalcia. Změřte sérovou koncentraci kalcia během 2 až 5 dnů před podáním dávky. Pokud je sérová hladina kalcia před dávkou pod 1,87 mmol/l nebo nad 2,55 mmol/l, mělo by se měření opakovat následující den. Upravte dávku aktivního vitamínu D nebo doplňku kalcia nebo obou na základě sérové hladiny kalcia a klinického hodnocení. Úpravy se mají opakovat, dokud nebudou cílové hladiny sérového kalcia před dávkou v rozsahu 2,0 – 2,25 mmol/l, aktivní vitamín D bude ukončen a doplněk kalcia je dostatečný pro splnění denních požadavků. Během titrace musí být monitorována koncentrace sérového kalcia. Dávka přípravku Natpar se může zvýšit o 25 mikrogramů přibližně každé 2 až 4 týdny, do maximální denní dávky 100 mikrogramů. K titraci dávky dolů na minimum 25 mikrogramů může dojít kdykoliv. Pokyny k úpravě dávky po úvodním období – viz SPC. Přípravek Natpar je vhodný pro podávání samotným pacientem jako subkutánní injekce jednou denně střídavě do každého stehna. Kontraindikace: Hypersenzitivita na léčivou látku nebo na kteroukoli pomocnou látku. U pacientů, kteří podstupují nebo dříve podstupovali radioterapii skeletu, s malignitami skeletu nebo kostními metastázami, kteří mají zvýšené výchozí riziko vzniku osteosarkomu (pacienti s Pagetovou chorobou kostí nebo dědičnými poruchami), s nevysvětleným zvýšením kostní alkalické fosfatázy a s pseudohypoparathyreózou. Zvláštní upozornění a opatření pro použití: Monitorování pacientů během léčby: Sérové hladiny kalcia před dávkou a v některých případech po dávce musí být během léčby přípravkem Natpar sledovány. Hyperkalcemie: často se objevovala během titračního období. V případě závažné hyperkalcemie (> 3,0 mmol/l nebo nad horní hranici normy s příznaky), zvažte hydrataci a dočasné zastavení podávání přípravku Natpar, kalcia a aktivního vitamínu D, dokud se sérové kalcium nevrátí do normálního rozsahu. Hypokalcemie: Riziko závažné hypokalcemie je nejvyšší po odložení podání, vynechání nebo náhlém vysazení přípravku Natpar, může se ale vyskytnout kdykoli. Souběžné použití se srdečními glykosidy: Hyperkalcemie z jakékoliv příčiny může predisponovat k toxicitě digitalis. Závažná porucha funkce ledvin nebo jater: Používat s opatrností u pacientů se závažnou poruchou ledvin nebo jater. Použití u mladých dospělých: Používat s opatrností u mladých dospělých pacientů s otevřenými epifýzami, protože tito pacienti mohou být vystaveni zvýšenému riziku vzniku osteosarkomu. Použití u starších pacientů: Klinické studie nezahrnovaly dostatečný počet subjektů ve věku ≥ 65 let pro stanovení, zda je odpověď u těchto subjektů odlišná od mladších subjektů. Tachyfylaxe: Účinek přípravku Natpar se může po čase u některých pacientů snížit. Urolitiáza: U pacientů s aktivní nebo nedávnou urolitiázou je nutno používat přípravek Natpar s opatrností vzhledem k jeho potenciálu tento stav zhoršovat. Hypersenzitivita: Po uvedení na trh byly hlášeny hypersenzitivní reakce, které mohou zahrnovat anafylaxi, dyspnoi, angioedém, kopřivku, vyrážku atd. Interakce: Kombinované použití přípravku Natpar a srdečních glykosidů může pacienty predisponovat k toxicitě digitalisu, pokud vznikne hyperkalcémie. Současné používání přípravku Natpar s bisfosfonáty se nedoporučuje. Fertilita, těhotenství a kojení: Údaje o podávání přípravku Natpar těhotným ženám nejsou k dispozici. Studie reprodukční toxicity na zvířatech nenaznačují přímé nebo nepřímé škodlivé účinky. Není známo, zda se přípravek Natpar vylučuje do lidského mateřského mléka. Dostupné farmakologické údaje u zvířat prokázaly vylučování přípravku Natpar do mléka. Neexistují žádné údaje u člověka o vlivu přípravku Natpar na plodnost. Studie na zvířatech nenaznačují žádnou poruchu plodnosti. Nežádoucí účinky: Nejčastější nežádoucí reakce byly hyperkalcemie, hypokalcemie a jejich související klinické projevy, zahrnující bolesti hlavy, průjem, zvracení, parestezie, hypestezie a hyperkalciurie. Ostatní nežádoucí účinky – viz SPC. Předávkování: Může způsobit hyperkalcémii. Těžká hyperkalcémie může být život ohrožující stav vyžadující neodkladnou lékařskou péči a pečlivé monitorování. Podmínky uchovávání: Uchovávejte v chladničce (2 °C – 8 °C). Chraňte před mrazem. Uchovávejte zásobní vložky v držáku v krabičce, aby byly chráněny před světlem. Podmínky uchovávání po rekonstituci – viz SPC. Držitel rozhodnutí o registraci: Shire Pharmaceuticals Ireland Limited, Block 2 & 3 Miesian Plaza, 50 – 58 Baggot Street Lower, Dublin 2, Irsko. Registrační čísla: EU/1/15/1078/001-004. Poslední revize SPC: 04/2020.
Výdej léčivého přípravku je vázán na lékařský předpis. Léčivý přípravek není hrazen z prostředků veřejného zdravotního pojištění. Před předepsáním se seznamte s úplným zněním Souhrnu údajů o přípravku.
Žádáme zdravotnické pracovníky, aby hlásili podezření na nežádoucí účinky na SÚKL nebo společnosti Takeda emailem na AE.CZE@takeda.com. Podezření na nežádoucí účinky hlaste také podle národních legislativních požadavků.
▼ Tento léčivý přípravek podléhá dalšímu sledování. To umožní rychlé získání nových informací o bezpečnosti. Žádáme zdravotnické pracovníky, aby hlásili jakákoli podezření na nežádoucí účinky. Podrobnosti o hlášení nežádoucích účinků viz bod 4.8 SPC.
Název přípravku: Natpar 25 mikrogramů/dávka prášek a rozpouštědlo pro injekční roztok; Natpar 50 mikrogramů/dávka prášek a rozpouštědlo pro injekční roztok; Natpar 75 mikrogramů/dávka prášek a rozpouštědlo pro injekční roztok; Natpar 100 mikrogramů/dávka prášek a rozpouštědlo pro injekční Složení: Jedna dávka obsahuje po rekonstituci 25/50/75/100 mikrogramů hormonum parathyroidum (rDNA)* v 71,4 mikrolitrech roztoku. Jedna zásobní vložka obsahuje 350/700/1050/1400 mikrogramů hormonum parathyroidum (rDNA). *Parathormon (rDNA) produkovaný E. coli pomocí rekombinantní DNA technologie je identický se sekvencí 84 aminokyselin endogenního lidského parathormonu. Indikace: Přípravek Natpar je indikován jako přídatná léčba dospělých pacientů s chronickou hypoparathyreózou, kteří nejsou dostatečně kontrolováni samostatnou standardní léčbou. Dávkování a způsob podání: Zahajte léčbu 50 mikrogramy jednou denně subkutánní injekcí do stehna (střídejte stehna každý den). Pokud je před podáním dávky sérové kalcium > 2,25 mmol/l, je možné zvážit podání počáteční dávky 25 mikrogramů. U pacientů užívajících aktivní vitamin D snižte jeho dávku o 50 %, pokud je sérové kalcium před podáním dávky nad 1,87 mmol/l. Udržujte doplňkovou dávku kalcia. Změřte sérovou koncentraci kalcia během 2 až 5 dnů před podáním dávky. Pokud je sérová hladina kalcia před dávkou pod 1,87 mmol/l nebo nad 2,55 mmol/l, mělo by se měření opakovat následující den. Upravte dávku aktivního vitamínu D nebo doplňku kalcia nebo obou na základě sérové hladiny kalcia a klinického hodnocení. Úpravy se mají opakovat, dokud nebudou cílové hladiny sérového kalcia před dávkou v rozsahu 2,0 – 2,25 mmol/l, aktivní vitamín D bude ukončen a doplněk kalcia je dostatečný pro splnění denních požadavků. Během titrace musí být monitorována koncentrace sérového kalcia. Dávka přípravku Natpar se může zvýšit o 25 mikrogramů přibližně každé 2 až 4 týdny, do maximální denní dávky 100 mikrogramů. K titraci dávky dolů na minimum 25 mikrogramů může dojít kdykoliv. Pokyny k úpravě dávky po úvodním období – viz SPC. Přípravek Natpar je vhodný pro podávání samotným pacientem jako subkutánní injekce jednou denně střídavě do každého stehna. Kontraindikace: Hypersenzitivita na léčivou látku nebo na kteroukoli pomocnou látku. U pacientů, kteří podstupují nebo dříve podstupovali radioterapii skeletu, s malignitami skeletu nebo kostními metastázami, kteří mají zvýšené výchozí riziko vzniku osteosarkomu (pacienti s Pagetovou chorobou kostí nebo dědičnými poruchami), s nevysvětleným zvýšením kostní alkalické fosfatázy a s pseudohypoparathyreózou. Zvláštní upozornění a opatření pro použití: Monitorování pacientů během léčby: Sérové hladiny kalcia před dávkou a v některých případech po dávce musí být během léčby přípravkem Natpar sledovány. Hyperkalcemie: často se objevovala během titračního období. V případě závažné hyperkalcemie (> 3,0 mmol/l nebo nad horní hranici normy s příznaky), zvažte hydrataci a dočasné zastavení podávání přípravku Natpar, kalcia a aktivního vitamínu D, dokud se sérové kalcium nevrátí do normálního rozsahu. Hypokalcemie: Riziko závažné hypokalcemie je nejvyšší po odložení podání, vynechání nebo náhlém vysazení přípravku Natpar, může se ale vyskytnout kdykoli. Souběžné použití se srdečními glykosidy: Hyperkalcemie z jakékoliv příčiny může predisponovat k toxicitě digitalis. Závažná porucha funkce ledvin nebo jater: Používat s opatrností u pacientů se závažnou poruchou ledvin nebo jater. Použití u mladých dospělých: Používat s opatrností u mladých dospělých pacientů s otevřenými epifýzami, protože tito pacienti mohou být vystaveni zvýšenému riziku vzniku osteosarkomu. Použití u starších pacientů: Klinické studie nezahrnovaly dostatečný počet subjektů ve věku ≥ 65 let pro stanovení, zda je odpověď u těchto subjektů odlišná od mladších subjektů. Tachyfylaxe: Účinek přípravku Natpar se může po čase u některých pacientů snížit. Urolitiáza: U pacientů s aktivní nebo nedávnou urolitiázou je nutno používat přípravek Natpar s opatrností vzhledem k jeho potenciálu tento stav zhoršovat. Hypersenzitivita: Po uvedení na trh byly hlášeny hypersenzitivní reakce, které mohou zahrnovat anafylaxi, dyspnoi, angioedém, kopřivku, vyrážku atd. Interakce: Kombinované použití přípravku Natpar a srdečních glykosidů může pacienty predisponovat k toxicitě digitalisu, pokud vznikne hyperkalcémie. Současné používání přípravku Natpar s bisfosfonáty se nedoporučuje. Fertilita, těhotenství a kojení: Údaje o podávání přípravku Natpar těhotným ženám nejsou k dispozici. Studie reprodukční toxicity na zvířatech nenaznačují přímé nebo nepřímé škodlivé účinky. Není známo, zda se přípravek Natpar vylučuje do lidského mateřského mléka. Dostupné farmakologické údaje u zvířat prokázaly vylučování přípravku Natpar do mléka. Neexistují žádné údaje u člověka o vlivu přípravku Natpar na plodnost. Studie na zvířatech nenaznačují žádnou poruchu plodnosti. Nežádoucí účinky: Nejčastější nežádoucí reakce byly hyperkalcemie, hypokalcemie a jejich související klinické projevy, zahrnující bolesti hlavy, průjem, zvracení, parestezie, hypestezie a hyperkalciurie. Ostatní nežádoucí účinky – viz SPC. Předávkování: Může způsobit hyperkalcémii. Těžká hyperkalcémie může být život ohrožující stav vyžadující neodkladnou lékařskou péči a pečlivé monitorování. Podmínky uchovávání: Uchovávejte v chladničce (2 °C – 8 °C). Chraňte před mrazem. Uchovávejte zásobní vložky v držáku v krabičce, aby byly chráněny před světlem. Podmínky uchovávání po rekonstituci – viz SPC. Držitel rozhodnutí o registraci: Shire Pharmaceuticals Ireland Limited, Block 2 & 3 Miesian Plaza, 50 – 58 Baggot Street Lower, Dublin 2, Irsko. Registrační čísla: EU/1/15/1078/001-004. Poslední revize SPC: 04/2020.
Výdej léčivého přípravku je vázán na lékařský předpis. Léčivý přípravek není hrazen z prostředků veřejného zdravotního pojištění. Před předepsáním se seznamte s úplným zněním Souhrnu údajů o přípravku.
Žádáme zdravotnické pracovníky, aby hlásili podezření na nežádoucí účinky na SÚKL nebo společnosti Takeda emailem na AE.CZE@takeda.com. Podezření na nežádoucí účinky hlaste také podle národních legislativních požadavků.
ADVATE
Zkrácené informace o léčivém přípravku
ADVATE 250 IU prášek a rozpouštědlo pro injekční roztok, ADVATE 500 IU prášek a rozpouštědlo pro injekční roztok, ADVATE 1000 IU prášek a rozpouštědlo pro injekční roztok, ADVATE 1500 IU prášek a rozpouštědlo pro injekční roztok, ADVATE 2000 IU prášek a rozpouštědlo pro injekční roztok, ADVATE 3000 IU prášek a rozpouštědlo pro injekční roztok
Dříve než začnete přípravek předepisovat, seznamte se, prosím, s úplným souhrnem údajů o přípravku (SPC).
Složení: Léčivá látka: Prášek: 250/500/1000/1500/2000/3000 IU lidského koagulačního faktoru VIII (rDNA), octocogum alfa, Rozpouštědlo 5 ml: Po rekonstituci obsahuje přípravek ADVATE přibližně 50/100/200/300/400/600 IU/ml lidského koagulačního faktoru VIII (rDNA), octocogum alfa.
Prášek 250/500/1000/1500 IU lidského koagulačního faktoru VIII (rDNA), octocogum alfa, Rozpouštědlo 2 ml: Po rekonstituci obsahuje přípravek ADVATE přibližně 125/250/500/750 IU/ml lidského koagulačního faktoru VIII (rDNA), octocogum alfa.
Indikace: Léčba a profylaxe krvácení u pacientů s hemofilií A (vrozený nedostatek faktoru VIII).
Dávkování a způsob podání: Léčba on demand (dle potřeby): Výpočet požadované dávky faktoru VIII je založen na empirickém zjištění, že 1 IU faktoru VIII na 1 kg tělesné hmotnosti zvyšuje aktivitu plazmatického faktoru VIII o 2 IU/dl. Dávka se určuje podle následujícího vzorce: Požadované jednotky (IU) = tělesná hmotnost (kg) x požadovaný vzestup faktoru VIII (%) x 0,5. Profylaxe: Pro dlouhodobou profylaxi krvácení u pacientů s těžkou hemofilií A jsou obvyklé dávky 20 až 40 IU faktoru VIII na 1 kg tělesné hmotnosti v intervalu dvou až tří dnů. U pacientů mladších 6 let se k profylaktické léčbě doporučují dávky faktoru VIII 20 až 50 IU na kg tělesné hmotnosti 3krát až 4krát týdně. Způsob podání: ADVATE má být podáván intravenózně. Kontraindikace: Hypersenzitivita na léčivou látku nebo na kteroukoli pomocnou látku nebo na myší nebo křeččí proteiny.
Upozornění: Hypersenzitivita: U přípravku ADVATE byly hlášeny hypersenzitivní reakce alergického typu (včetně anafylaxe). Přípravek obsahuje stopy myších a křeččích proteinů. Pokud se symptomy hypersenzitivity objeví, pacienti musí ihned přerušit používání přípravku a kontaktovat svého lékaře. Inhibitory: Tvorba neutralizujících protilátek (inhibitorů) faktoru VIII je známou komplikací léčby jedinců s hemofilií A. Riziko vzniku inhibitorů souvisí se závažností onemocnění i s expozicí faktoru VIII, přičemž toto riziko je nejvyšší během prvních 20 dnů expozice. Obecně platí, že všichni pacienti léčení přípravky s koagulačním faktorem VIII musí být pečlivě sledováni s ohledem na vznik inhibitorů pomocí příslušných klinických pozorování a laboratorních testů. Důrazně se doporučuje po každém podání přípravku ADVATE pacientovi zaznamenat název a číslo šarže přípravku, aby bylo možné přesně dohledat, jaká šarže léčivého přípravku byla pacientovi podána.
Významné interakce: Nebyly provedeny žádné studie interakce s přípravkem ADVATE. Studie kompatibility nejsou k dispozici, a proto nesmí být tento léčivý přípravek mísen s jinými léčivými přípravky nebo rozpouštědly. Hlavní nežádoucí účinky: Klinické studie s přípravkem ADVATE zahrnovaly 418 pacientů s alespoň jednou expozicí přípravku ADVATE; bylo při nich nahlášeno celkem 93 nežádoucích účinků léčiva (NÚ). NÚ, které se vyskytovaly s nejvyšší frekvencí, byly vývoj neutralizujících protilátek faktoru VIII (inhibitorů), bolest hlavy a horečka. Alergické reakce nebo hypersenzitivní reakce byly pozorovány vzácně a mohou v některých případech progredovat do těžké anafylaxe (včetně šoku). Může být pozorován vývoj protilátek proti myšímu a/nebo křeččímu proteinu se souvisejícími hypersenzitivními reakcemi. K rozvoji neutralizujících protilátek (inhibitorů) může dojít u pacientů s hemofilií A, kteří jsou léčeni faktorem VIII, včetně přípravku ADVATE. Pokud se takové inhibitory objeví, projeví se tento stav jako nedostatečná klinická odpověď.
Uchovávání: Uchovávejte v chladničce (2°C – 8°C). Chraňte před mrazem. Během doby použitelnosti může být přípravek uchováván při pokojové teplotě (do 25°C) po jedno období nepřesahující 6 měsíců. Přípravek nesmí být vrácen zpět do chladničky.
Držitel rozhodnutí o registraci: Takeda Manufacturing Austria AG, Industriestrasse 67, A-1221 Vídeň, Rakousko.
Registrační čísla: EU/1/03/271/001-020.
Poslední revize SPC: 02/2021.
Výdej léčivého přípravku je vázán na lékařský předpis.
Léčivý přípravek je hrazen z prostředků veřejného zdravotního pojištění.
Úplné znění SPC naleznete na www.sukl.cz.
Žádáme zdravotnické pracovníky, aby hlásili podezření na nežádoucí účinky na SÚKL nebo společnosti Takeda emailem na AE.CZE@takeda.com. Podezření na nežádoucí účinky hlaste také podle národních legislativních požadavků.
Dříve než začnete přípravek předepisovat, seznamte se, prosím, s úplným souhrnem údajů o přípravku (SPC).
Složení: Léčivá látka: Prášek: 250/500/1000/1500/2000/3000 IU lidského koagulačního faktoru VIII (rDNA), octocogum alfa, Rozpouštědlo 5 ml: Po rekonstituci obsahuje přípravek ADVATE přibližně 50/100/200/300/400/600 IU/ml lidského koagulačního faktoru VIII (rDNA), octocogum alfa.
Prášek 250/500/1000/1500 IU lidského koagulačního faktoru VIII (rDNA), octocogum alfa, Rozpouštědlo 2 ml: Po rekonstituci obsahuje přípravek ADVATE přibližně 125/250/500/750 IU/ml lidského koagulačního faktoru VIII (rDNA), octocogum alfa.
Indikace: Léčba a profylaxe krvácení u pacientů s hemofilií A (vrozený nedostatek faktoru VIII).
Dávkování a způsob podání: Léčba on demand (dle potřeby): Výpočet požadované dávky faktoru VIII je založen na empirickém zjištění, že 1 IU faktoru VIII na 1 kg tělesné hmotnosti zvyšuje aktivitu plazmatického faktoru VIII o 2 IU/dl. Dávka se určuje podle následujícího vzorce: Požadované jednotky (IU) = tělesná hmotnost (kg) x požadovaný vzestup faktoru VIII (%) x 0,5. Profylaxe: Pro dlouhodobou profylaxi krvácení u pacientů s těžkou hemofilií A jsou obvyklé dávky 20 až 40 IU faktoru VIII na 1 kg tělesné hmotnosti v intervalu dvou až tří dnů. U pacientů mladších 6 let se k profylaktické léčbě doporučují dávky faktoru VIII 20 až 50 IU na kg tělesné hmotnosti 3krát až 4krát týdně. Způsob podání: ADVATE má být podáván intravenózně. Kontraindikace: Hypersenzitivita na léčivou látku nebo na kteroukoli pomocnou látku nebo na myší nebo křeččí proteiny.
Upozornění: Hypersenzitivita: U přípravku ADVATE byly hlášeny hypersenzitivní reakce alergického typu (včetně anafylaxe). Přípravek obsahuje stopy myších a křeččích proteinů. Pokud se symptomy hypersenzitivity objeví, pacienti musí ihned přerušit používání přípravku a kontaktovat svého lékaře. Inhibitory: Tvorba neutralizujících protilátek (inhibitorů) faktoru VIII je známou komplikací léčby jedinců s hemofilií A. Riziko vzniku inhibitorů souvisí se závažností onemocnění i s expozicí faktoru VIII, přičemž toto riziko je nejvyšší během prvních 20 dnů expozice. Obecně platí, že všichni pacienti léčení přípravky s koagulačním faktorem VIII musí být pečlivě sledováni s ohledem na vznik inhibitorů pomocí příslušných klinických pozorování a laboratorních testů. Důrazně se doporučuje po každém podání přípravku ADVATE pacientovi zaznamenat název a číslo šarže přípravku, aby bylo možné přesně dohledat, jaká šarže léčivého přípravku byla pacientovi podána.
Významné interakce: Nebyly provedeny žádné studie interakce s přípravkem ADVATE. Studie kompatibility nejsou k dispozici, a proto nesmí být tento léčivý přípravek mísen s jinými léčivými přípravky nebo rozpouštědly. Hlavní nežádoucí účinky: Klinické studie s přípravkem ADVATE zahrnovaly 418 pacientů s alespoň jednou expozicí přípravku ADVATE; bylo při nich nahlášeno celkem 93 nežádoucích účinků léčiva (NÚ). NÚ, které se vyskytovaly s nejvyšší frekvencí, byly vývoj neutralizujících protilátek faktoru VIII (inhibitorů), bolest hlavy a horečka. Alergické reakce nebo hypersenzitivní reakce byly pozorovány vzácně a mohou v některých případech progredovat do těžké anafylaxe (včetně šoku). Může být pozorován vývoj protilátek proti myšímu a/nebo křeččímu proteinu se souvisejícími hypersenzitivními reakcemi. K rozvoji neutralizujících protilátek (inhibitorů) může dojít u pacientů s hemofilií A, kteří jsou léčeni faktorem VIII, včetně přípravku ADVATE. Pokud se takové inhibitory objeví, projeví se tento stav jako nedostatečná klinická odpověď.
Uchovávání: Uchovávejte v chladničce (2°C – 8°C). Chraňte před mrazem. Během doby použitelnosti může být přípravek uchováván při pokojové teplotě (do 25°C) po jedno období nepřesahující 6 měsíců. Přípravek nesmí být vrácen zpět do chladničky.
Držitel rozhodnutí o registraci: Takeda Manufacturing Austria AG, Industriestrasse 67, A-1221 Vídeň, Rakousko.
Registrační čísla: EU/1/03/271/001-020.
Poslední revize SPC: 02/2021.
Výdej léčivého přípravku je vázán na lékařský předpis.
Léčivý přípravek je hrazen z prostředků veřejného zdravotního pojištění.
Úplné znění SPC naleznete na www.sukl.cz.
Žádáme zdravotnické pracovníky, aby hlásili podezření na nežádoucí účinky na SÚKL nebo společnosti Takeda emailem na AE.CZE@takeda.com. Podezření na nežádoucí účinky hlaste také podle národních legislativních požadavků.
Vše o hemofilii
Společnost Takeda je tradičním partnerem pacientů s hemofilií i zdravotníků, kteří o ně pečují. V České republice mohou hemofilici i lékaři profitovat z našich:
- dlouhodobých zkušeností,
- intenzivního výzkumu,
- moderních léčivých přípravků a technologií, které přinášíme na trh.
Zaměřujeme se také na optimalizaci léčby, zvyšování komfortu jejího podávání i spolupráci s odbornými společnostmi a pacientskými organizacemi. Jsme připraveni reagovat na výzvy, které léčba hematologických onemocnění přináší. V této oblasti disponujeme rozsáhlým portfoliem léčivých přípravků, zahrnujícím plazmatické, rekombinantní produkty i léčiva s prolongovaným působením.
Přinášíme sofistikovaná léčiva i pomůcky pro léčbu hemofilie všech typů a věříme, že se nám i s Vaší pomocí podaří naplnit naši vizi Bleed-Free World a že se „svět bez krvácení“ v budoucnu skutečně stane realitou. Vyzkoušejte se svými pacienty první spojení webové a mobilní aplikace v léčbě hemofilie a jediný schválený zdravotnický prostředek k individuálnímu nastavení optimální profylaxe hemofilika – webovou a mobilní aplikaci myPKFiT.
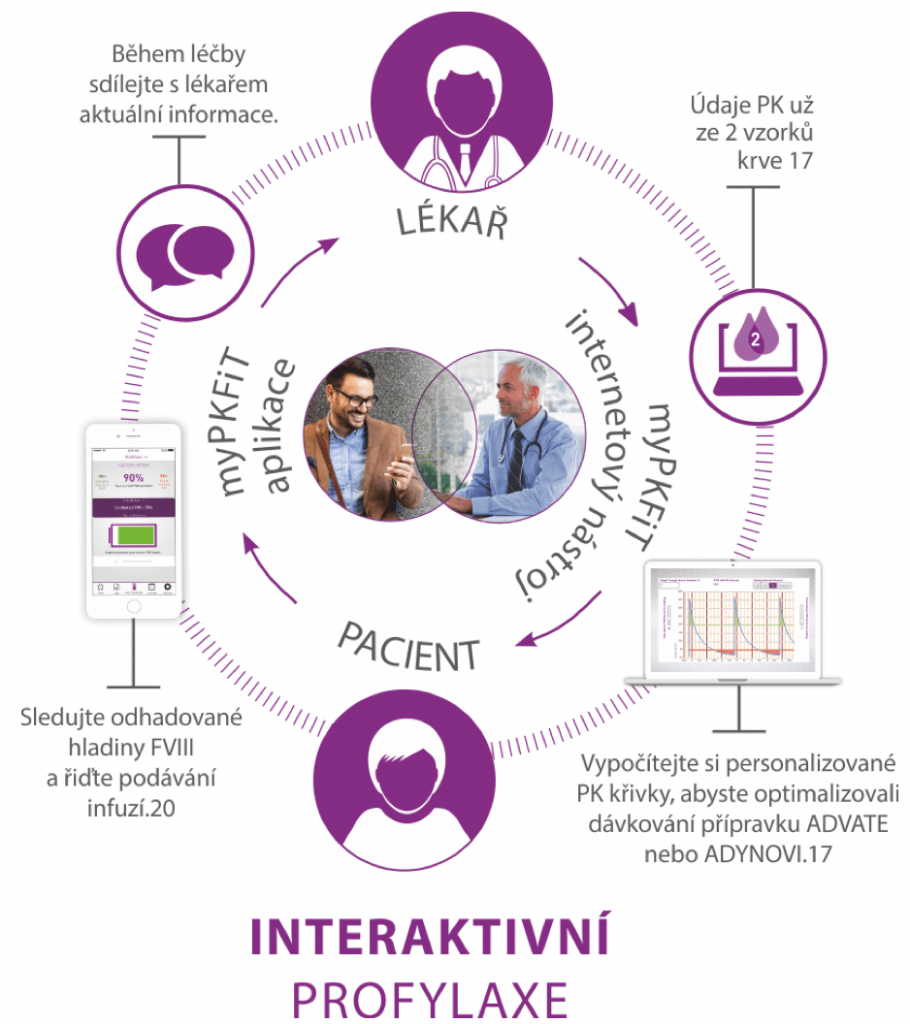
Používání aplikace myPKFiT umožňuje sdílet všechny aktualizace s ošetřujícím lékařem, optimalizovat dávkování na základě osobního farmakokinetického profilu a zároveň zvyšovat osobní účast na léčbě a samostatnost v jejím managementu. I v ČR se toto řešení setkalo s velmi příznivým ohlasem u odborníků i pacientů a velké množství z nich již výhod této technologie naplno využívá. Buďte dalšími z nich.
Stručně o hemofilii
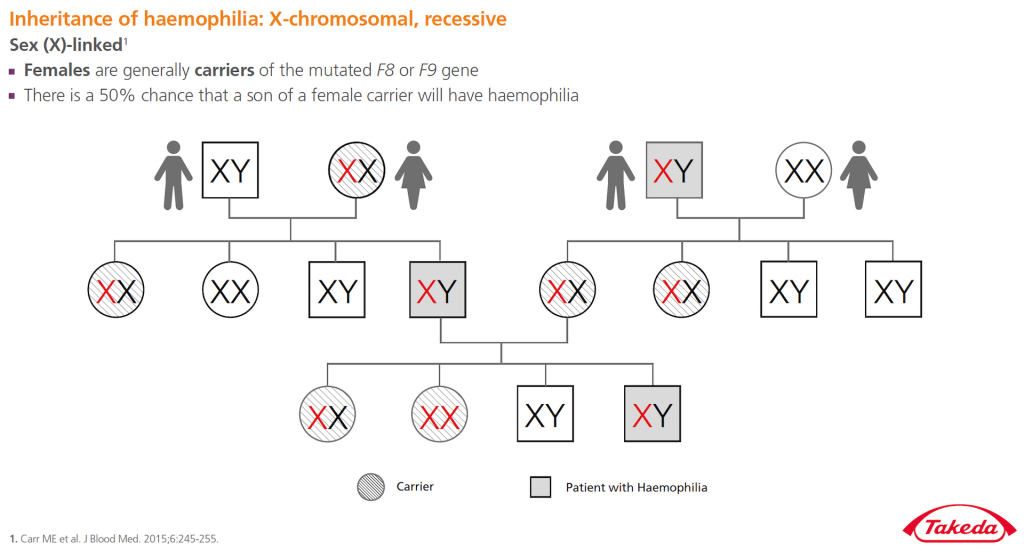
Hemofilie je vzácná, obvykle dědičná koagulopatie. Jedná se o gonosomálně recesivní onemocnění. Defektní gen je lokalizován na pohlavním chromosomu X, proto se hemofilie vyskytuje prakticky jen u mužů, u žen bývá extrémně vzácná. Hemofilie se projevuje poruchou srážlivosti krve a z toho plynoucím krvácením. Zdaleka nejčastějším typem je hemofilie typu A (deficit faktoru VIII). Druhým nejčastějším typem je typ B (deficit faktoru IX). Existují i jiná krvácivá onemocnění s deficity jiných srážecích faktorů, která se obvykle již jako hemofilie neoznačují.
Hemofilii lze odhalit již u batolat, kdy dítě začíná chodit a také padat. Ke krvácení dochází i při minimálním nárazu nebo zcela bez příčiny. Na exponovaných místech (hlava, hýždě, končetiny) se pak objevují typické podkožní a někdy i svalové hematomy. Po pádu na obličej nezřídka dochází
i ke krvácení v dutině ústní. Tyto projevy může okolí rodiny mylně vyhodnotit jako týrání dítěte.
V novorozeneckém věku je krvácení poměrně vzácné. U mladších dětí a školáků může frekvenci krvácení zesílit i stres, například v období zkoušek. Pozoruje se i sezónní vliv na krvácení, například na jaře a na podzim.
Hemofilii rozdělujeme v závislosti na množství chybějícího koagulačního faktoru na:
– Těžkou formu (< 1 IU/dl nebo < 1 % normální koncentrace srážecího faktoru), při které dochází k častým spontánním krvácením především do velkých kloubů nebo svalů.1
– Středně těžkou formu (1–5 % normální koncentrace srážecího faktoru). Krvácení vzniká již po nepatrných úrazech nebo poraněních, může vzniknout i spontánně.
– Lehkou formu (> 5–40 % normální koncentrace srážecího faktoru). Ke dlouhotrvajícímu krvácení dochází po stomatologických a operačních výkonech či úrazech, ale pacient nekrvácí spontánně do kloubů nebo dalších částí těla.1
C-APROM/CZ//0701
Literatura
- Srivastava A, Brewer AK, Mauser-Bunschoten EP, et al.; Treatment Guidelines Working Group on Behalf of The World Federation Of Hemophilia. Guidelines for the management of hemophilia. Haemophilia 2013; 19 (1): e1–e47.
- Blanchette VS, Key NS, Ljung LR, et al.; Subcommittee on Factor VIII, Factor IX and Rare Coagulation Disorders of the Scientific and Standardization Committee of the International Society on Thrombosis and Hemostasis. Definitions in hemophilia: communication from the SSC of the ISTH. J Thromb Haemost 2014; 12 (11): 1935–1939.
- Rodriguez-Merchan EC. Musculoskeletal complications of hemophilia. HSS J 2010; 6 (1): 37–42.
- Aledort LM, Haschmeyer RH, Pettersson H. A longitudinal study of orthopaedic outcomes for severe factor-VIII deficient haemophiliacs. The Orthopaedic Outcome Study Group. J Intern Med 1994; 236 (4): 391–399.
- National Hemophilia Foundation. MASAC recommendation concerning prophylaxis (regular administration of clotting factor concentrate to prevent bleeding). MASAC Document #241. February 28, 2016.
- Oldenburg J, Zimmermann R, Katsarou O, et al. Controlled, cross-sectional MRI evaluation of joint status in severe haemophilia A patients treated with prophylaxis vs. on demand. Haemophilia 2015; 21 (2): 171–179.
- Pasca S, Milan M, Sarolo L, Zanon E. PK-driven prophylaxis versus standard prophylaxis: When a tailored treatment may be a real and achievable cost-saving approach in children with severe hemophilia A. Thromb Res 2017; 157: 58–63.
- Údaje z prospektivní klinické studie fáze IV s Advate (n = 66).
- Iorio A, Marcucci M, Cheng J, et al. Patient data meta-analysis of Post-Authorization Safety Surveillance (PASS) studies of haemophilia A patients treated with rAHF-PFM. Haemophilia 2014; 20 (6): 777–783.
- Pět poregistračních studií (PASS) s Advate.
- Mahdi AJ, Obaji SG, Collins PW. Role of enhanced halife factor VIII and IX in the treatment of haemophilia. Br J Haematol 2015; 169 (6): 768–776.
- Konkle BA, Stasyshyn O, Chowdary P, et al. Pegylated, full-length, recombinant factor VIII for prophylactic and on-demand treatment of severe hemophilia A. Blood 2015; 126 (9): 1078–1085.
- Valentino LA. Considerations in individualizing prophylaxis in patients with haemophilia A. Haemophilia 2014; 20 (5): 607–615.
- Windyga J, Zbikowski P, Ambroziak P, et al. Management of factor VII-deficient patients undergoing joint surgeries–preliminary results of locally developed treatment regimen. Haemophilia 2013; 19 (1): 89–93.
- Urasinski T, Stasyshyn O, Andreeva T, et al. Recombinant factor IX (BAX326) in previously treated paediatric patients with haemophilia B: a prospective clinical trial. Haemophilia 2015; 21 (2): 196–203.
- Gouw SC, van der Bom JG, van den Berget HM. Treatment-related risk factors of inhibitor development in previously untreated patients with hemophilia A: the CANAL cohort study. Blood 2007; 109 (11): 4648–4654.
- Coppola A, Di Capua M, Di Minno MN, et al. Treatment of hemophilia: a review of current advances and ongoing issues. J Blood Med 2010; 1: 183–195.
- Morfini M, Auerswald G, Kobelt RA, et al. Prophylactic treatment of haemophilia patients with inhibitors: clinical experience with recombinant factor VIIa in European Haemophilia Centres. Haemophilia 2007; 13 (5): 502–507.
- Doc. MUDr. Zuzana Čermáková, Ph.D. – Centrum pro trombózu a hemostázu, hemofilické centrum, Krevní centrum FN Ostrava, Farmakologický ústav, LF MU v Brně.
- Čermáková Z, Hrdličková R, Smejkal P, et al.; Pracovní skupina ČNHP pro standardy. Diagnostika a léčba získané hemofilie – konsenzuální doporučení Českého národního hemofilického programu (ČNHP). Transfuze Hematol dnes 2017; 23 (2): 101–109.