
Oblast primární a sekundární imunodeficience
Při primární (vrozené) imunodeficienci (PID) se v těle nevytvářejí v dostatečné míře některé součásti imunitního systému (imunoglobuliny – Ig, T-lymfocyty, fagocyty, složky komplementu) nebo některé z nich nefungují správně. Jedná se o více než 400 chronických poruch, vznikajících v důsledku genetické vady nebo změn v lidské DNA, v jejichž důsledku může u postiženého jedince existovat zvýšená náchylnost k bakteriálním a virovým infekcím (i atypickými agens – oportunními patogeny) a také vyšší nemocnost (banálními infekcemi i pneumoniemi, sinusitidami, meningitidami a abscesy), přičemž infekce většinou špatně odpovídá na konvenční léčbu. V Evropě trpí PID cca 10 000 osob a v převážné míře se manifestuje jako deficience protilátková, případně jako deficit T-buněk (a kombinované poruchy). Tato patologie se u jedince může objevit, aniž by se PID vyskytovala v rodinné anamnéze.
Pacientovi s imunodeficitem je nutno v rámci substituční imunoglobulinové terapie podávat infuzí nebo injekčně (intravenózně či subkutánně) protilátky získané z krve zdravých jedinců. Další možností léčby je transplantace kostní dřeně či krvetvorných buněk a genová terapie.
Sekundární (získané) imunodeficience (SID) nejsou až tak vzácné jako PID. Nevznikají na podkladě genetické poruchy, nýbrž podvýživy, jiné nemoci (chronická lymfatická leukémie – CLL, mnohočetný myelom – MM) nebo intervence, dysregulací imunitního systému při napadení tkáně hostitelského organismu, často prostřednictvím složitých mechanismů (většinou u neuromuskulárního postižení), a jsou známy i jako polékové reakce (při systémové kortikoterapii, imunosupresivní léčbě a protinádorové chemoterapii či radioterapii). Iatrogenní imunosuprese se také zaznamenává v souvislosti s některými pooperačními stavy (u popálenin nebo po splenektomii). Nejčastějším projevem sekundárních imunodeficiencí jsou rekurentní infekce v oblasti dýchacích cest a trávicího traktu. Při klinických projevech hypogamaglobulinemie má léčba charakter substituční terapie imunoglobuliny.
Vedle nitrožilního (i.v.) podání imunoglobulinů (IVIg) se používají také subkutánní (s.c.) aplikace (SCIg). K dispozici jsou i sofistikované produkty (facilitované přípravky – fSCIg), využívající dočasného zvýšení přirozeného obratu hyaluronanu v kůži s otevřením mikroskopických kanálků v subkutánní tkáni, což zvyšuje její propustnost.1 Podkožní podání také zvyšuje komfort léčby – současným trendem je rychlý přesun pacienta do domácího léčení a po zaškolení i domácí podávání Ig.
V této oblasti jsou Vám k dispozici tyto produkty společnosti Takeda:
![]()
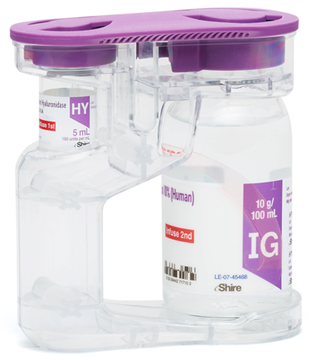
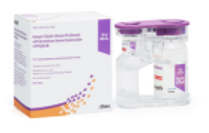
HyQvia je první a jediný SCIg s rekombinantní lidskou hyaluronidázou, která po podkožním podání zvyšuje rozptyl a vstřebávání imunoglobulinů. Má charakter facilitovaného 10% SCIg k substituční terapii dospělých, dětí i dospívajících (0–18 let), nevyžaduje žilní přístup a umožňuje podat najednou celou měsíční dávku Ig (obvykle na jednom infuzním místě). Má 93% biologickou dostupnost a režim podávání lze upravit dle léčebných potřeb pacienta. Příznivým faktem je, že při této léčbě se objevuje méně systémových nežádoucích účinků než u IVIg (8 % vs. 25 %), méně nežádoucích účinků za měsíc než u konvenčních SCIg produktů a že bylo zaznamenáno 2,97 infekcí na pacienta/rok oproti 4,51 při terapii IVIg a jen 0,025 akutních závažných bakteriálních infekcí za pacientorok. Většina (98,7 %) nežádoucích účinků byla považována za mírné nebo středně závažné.2
2 Wasserman RL, Melamed I, Stein MR, et al. Recombinant human hyaluronidase-facilitated subcutaneous infusion of human immunoglobulins for primary immunodeficiency. J Allergy Clin Immunol 2012; 130:951–957.e11.
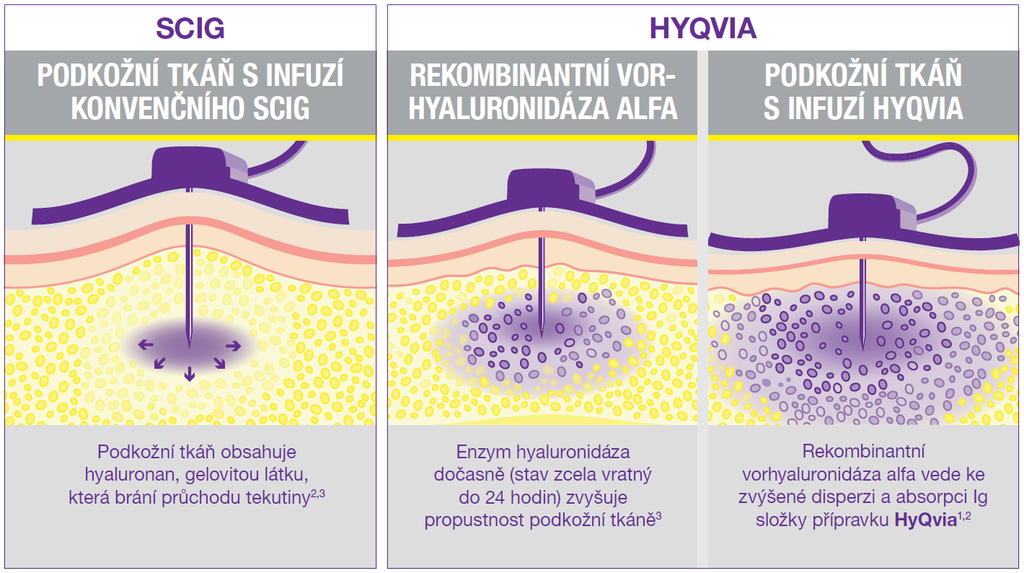
2 Wasserman R. L. et al. J Allergy Clin Immunol. 2012; 130 (4): 951–957.
3 Bookbinder L. H. et al. J Control Release. 2006; 114 (2): 230–241.
HyQvia – facilitovaný podkožně podávaný imunoglobulin (fSCIG)
Rekombinantní Ulehčuje disperzi a absorbci IgG – umožňuje podání vyššího objemu SCIG do místa aplikace |
 |
Normální lidský imunoglobulín 10%: Nositel terapeutického účinku |
IG terapie, která umožňuje aplikaci celé měsíční dávky*
1 jehlou na 1 infuzní místo** 1 x měsíčně a to v domácím prostředí.
Souhrn údajů o přípravku HyQvia (SPC), září 2021, Blau I-W, et al. Expert Rev Clin Immunol 2016;13:1–7.
*každé 3-4 týdny, **Frekvence infuzí může být upravena podle klinické odpovědi pacienta
![]()
Cuvitru je normální lidský imunoglobulin (SCIg) 20% s podáváním 1× týdně, který někteří pacienti preferují. Francouzská studie ukázala, že co se týká interference při léčbě, jsou pacienti s produkty pro domácí aplikaci SCIg výrazně spokojenější, než je tomu u IVIg, a jsou i spokojenější s SCIg produkty oproti substituci IVIg podávané v nemocnici.1 Potěšitelné je, že u terapie Cuvitru 2 ze 3 pacientů nevykazují lokální nežádoucí účinky a 99,8 % hodnocených infuzí bylo dokončeno bez přerušení, snížení rychlosti nebo ukončení z důvodu nesnášenlivosti.2,3 Výhodou je i to, že Cuvitru umožňuje podání do < 1 hodiny (až 60 ml/h/místo).
2 Borte M, Kriván G, Derfalvi B, et al. Efficacy, safety, tolerability and pharmacokinetics of a novel human immune globulin subcutaneous, 20%: a phase 2/3 study in Europe in patients with primary immunodeficiencies. Clin Exp Immunol 2017;187:146–159.
3 Suez D, Stein M, Gupta S, et al. Efficacy, safety, and pharmacokinetics of a novel human immune globulin subcutaneous, 20 % in patients with primary immunodeficiency diseases in North America. J Clin Immunol 2016;36:700–712.
4 Souhrn údajů o přípravku Cuvitru (SPC), září 2021.
![]()
Kiovig je normální lidský imunoglobulin 10% k infuznímu podání. Je určen k substituční a imunomodulační terapii dospělých, dětí i dospívajících (0–18 let), především u syndromů primárního imunodeficitu s poruchou tvorby protilátek, sekundárních imunodeficitů u pacientů trpících závažnými nebo opakovanými infekcemi a také například u pacientů s Kawasakiho chorobou, chronickou zánětlivou demyelinizační polyradikuloneuropatií (CIDP), multifokální motorickou neuropatií (MMN) nebo s Guillain-Barrého syndromem. Jeho výhodou je 100% biologická dostupnost.1
Imunoglobuliny – specifika typů terapie
| Intravenózní IG (IVIG) |
Konvenční SCIG (cSCIG) |
Facilitovaný SCIG (fSCIG) |
||||
| Samostatné podání1,2,5 |
|
|
|
|||
| Typický počet míst podání na infuzi1,6 |
|
|
|
|||
| Objem na infuzi1,6 |
|
|
|
|||
| Frekvence infuzí1,6 |
|
|
|
|||
| Biologická dostupnost7 |
|
 |
|
 |
|
 |
| Celková doba podávání infuze za měsíc3,4,6 |
|
|
|
|||
1 Jolles S, et al. Clin Exp Immunol. 2015;179(2):146-160. 2 Jolles S. Immunotargets Ther. 2013;2:125-133. 3 Kirmse J. Home Healthc Nurse. 2009;27(2):104-111. 4 Skoda-Smith S, et al. Ther Clin Risk Manag. 2010;6:1-10. 5 Souhrn údajů přípravku HyQvia, září 2021 6 Wasserman RL, et al. J Allergy Clin Immunol. 2012;130(4):951-957. 7 Wasserman RL. Immunotherapy. 2014. 8 Borte M, Kriván G, Derfalvi B, et al. Efficacy, safety, tolerability and pharmacokinetics of a novel human immune globulin subcutaneous, 20%: a phase 2/3 study in Europe in patients with primary immunodeficiencies. Clin Exp Immunol 2017;187:146–159.
C-APROM/CZ/CUVI/0019 Srpen 2022



HyQvia 100 mg/ml – infuzní roztok k subkutánnímu podání
Zkrácené informace o přípravku
Dříve než začnete přípravek předepisovat, seznamte se, prosím, s úplným souhrnem údajů o přípravku (SPC).
▼ Tento léčivý přípravek podléhá dalšímu sledování. To umožní rychlé získání nových informací o bezpečnosti. Žádáme zdravotnické pracovníky, aby hlásili jakákoli podezření na nežádoucí účinky. Podrobnosti o hlášení nežádoucích účinků viz bod 4.8 SPC.
Složení: Jedna injekční lahvička immunoglobulinum humanum normale (IG 10%) a jedna injekční lahvička hyaluronidasum humanum biosyntheticum (rHuPH20). Léčivá látka: Jeden ml obsahuje 100 mg normálního lidského imunoglobulinu (o čistotě alespoň 98 % IgG). Maximální obsah IgA je 140 mikrogramů/ml. Pomocné látky: Rekombinantní lidská hyaluronidáza (rHuPH20) je purifikovaný glykoprotein o 447 aminokyselinách produkovaný buňkami vaječníku čínského křečíka (CHO) technologií rekombinantní DNA. Injekční lahvička normálního lidského imunoglobulinu (IG 10%): Glycin, voda pro injekci. Injekční lahvička rekombinantní lidské hyaluronidázy (rHuPH20): Chlorid sodný, hydrogenfosforečnan sodný, lidský albumin, dinatriumedetát (EDTA), chlorid vápenatý, hydroxid sodný (pro úpravu pH), kyselina chlorovodíková (pro úpravu pH), voda pro injekci.
Indikace: Substituční terapie dospělých, dětí a dospívajících (0–18 let) u syndromů primární imunodeficience s narušenou tvorbou protilátek, u hypogamaglobulinemie a rekurentních bakteriálních infekcí u pacientů s chronickou lymfatickou leukemií (CLL), u nichž selhala profylaktická antibiotika nebo jsou kontraindikována, u hypogamaglobulinemie a rekurentních bakteriálních infekcí u pacientů s mnohočetným myelomem (MM) a u hypogamaglobulinemie u pacientů před a po alogenní transplantaci hematopoetických kmenových buněk (HSCT).
Dávkování a způsob podání: Substituční terapie by měla být zahájena a monitorována lékařem zkušeným v léčbě imunodeficience. Dávka a dávkovací režim závisí na indikaci. Při substituční terapii je dávka u každého pacienta individuální, závisí na konkrétní farmakokinetice a klinické odpovědi. Následující dávkovací režimy jsou pouze orientační. Pacienti dosud neléčení imunoglobuliny: Dávka potřebná k dosažení minimální hladiny 6 g/l v rovnovážném stavu je řádově 0,4–0,8 g/kg/měsíc. Dávkovací interval k udržení hladin v rovnovážném stavu se liší v rozmezí 2–4 týdnů. Kumulativní měsíční dávka IG 10% by měla být rozdělena do jednotýdenních, dvoutýdenních atd. dávek podle naplánovaných léčebných intervalů přípravkem HyQvia. Pacienti dříve léčení imunoglobulinem podávaným intravenózně: Léčivý přípravek by měl být podáván ve stejné dávce a se stejnou četností jako jejich předchozí intravenózní imunoglobulinová léčba. Pokud byli dříve pacienti na 3týdenním dávkovacím režimu, zvýšení intervalu na 4 týdny lze dosáhnout podáváním stejných týdenních ekvivalentů. Pacienti dříve léčení subkutánně podávaným imunoglobulinem: Úvodní dávka přípravku HyQvia je stejná jako u subkutánní léčby, lze ji však přizpůsobit 3 až 4týdennímu intervalu. První infuze přípravku HyQvia by měla být podána jeden týden po poslední léčbě předchozím imunoglobulinem. Pediatrická populace: Dávkování u dětí a dospívajících (0–18 let) se neliší od dávkování u dospělých, neboť dávkování v každé indikaci je dáno tělesnou hmotností a upravuje se podle klinického výsledku výše zmíněného onemocnění. Způsob podání: pouze k subkutánnímu podání, nepodávejte intravenózně. Přípravek HyQvia je tvořen dvěma injekčními lahvičkami. Každá injekční lahvička IG 10% je dodávána s odpovídajícím množstvím rekombinantní lidské hyaluronidázy, viz informace v SPC.
Kontraindikace: HyQvia se nesmí podávat intravenózně ani intramuskulárně. Hypersenzitivita na léčivou látku (IgG) nebo na kteroukoli pomocnou látku. Hypersenzitivita na lidské imunoglobuliny, zejména ve velmi vzácných případech deficitu IgA, kdy má pacient proti IgA protilátky. Známá systémová hypersenzitivita na hyaluronidázu nebo rekombinantní lidskou hyaluronidázu.
Upozornění: Pokud je přípravek HyQvia náhodně aplikován do žíly, může u pacienta vyvolat šok. Používejte doporučené rychlosti infuze. Pacienty je nutné důsledně sledovat v průběhu celé infuze, a to především pacienty začínající s léčbou. Určité nežádoucí účinky se mohou objevovat častěji u pacientů, kteří dostávají normální lidský imunoglobulin poprvé nebo (ve vzácných případech) jej mění nebo pokud uběhla dlouhá doba od předchozí infuze. V případě nežádoucích účinků je nutné buď snížit rychlost podávání infuze, nebo ji úplně zastavit. V případě šoku okamžitě ukončete infuzi a zahajte u pacienta léčbu šoku. Hypersenzitivita na IG 10%: Pravé reakce přecitlivělosti jsou vzácné. Může k nim docházet především u pacientů s protilátkami proti IgA, které je třeba léčit se zvýšenou opatrností. Hypersenzitivita na rekombinantní lidskou hyaluronidázu: Jakékoli podezření na reakci podobnou alergické nebo anafylaktické reakci po podání rekombinantní lidské hyaluronidázy vyžaduje okamžité přerušení infuze a – podle konkrétní potřeby – zahájení standardního léčebného postupu. Imunogenicita rekombinantní lidské hyaluronidázy: U pacientů léčených přípravkem HyQvia v klinických studiích byl hlášen vznik jiných než neutralizačních protilátek proti rekombinantní lidské hyaluronidáze. Tromboembolismus: S použitím imunoglobulinů byly spojeny arteriální a venózní tromboembolické příhody, včetně infarktu myokardu, mozkové příhody, hluboké žilní trombózy a plicní embolie. Před použitím imunoglobulinů musí být pacienti dostatečně hydratováni. U pacientů s preexistujícími rizikovými faktory výskytu tromboembolické příhody je třeba postupovat s opatrností. Hemolytická anémie. Imunoglobulinové přípravky obsahují protilátky proti krevním skupinám (např. A, B, D), které se mohou chovat jako hemolyziny. Syndrom aseptické meningitidy (AMS): Ve spojení s intravenózní a subkutánní imunoglobulinovou léčbou byl hlášen výskyt syndromu aseptické meningitidy. AMS se může častěji objevovat ve spojitosti s vysokou dávkou (2 g/kg) intravenózní imunoglobulinovou léčbou. Důležité informace o některých složkách přípravku HyQvia: Přípravek HyQvia neobsahuje cukry. Složka IG 10% obsahuje stopová množství sodíku. Rekombinantní lidská hyaluronidáza obsahuje 4,03 mg sodíku na ml, s maximální denní dávkou přibližně 120 mg. Interference se sérologickými testy: Po imunoglobulinové infuzi může mít přechodný vzestup různých pasivně přenášených protilátek v krvi pacienta za následek zavádějící pozitivní výsledky sérologických testů. Přenosná agens: Normální lidský imunoglobulin a lidský sérový albumin (stabilizátor rekombinantní lidské hyaluronidázy) se vyrábějí z lidské plazmy. Standardní opatření zabraňující přenosu infekce zahrnují pečlivý výběr dárců, testování jednotlivých odběrů krve a plazmatických poolů na specifické ukazatele infekce a určité výrobní kroky, při nichž jsou inaktivovány nebo odstraněny viry. Přes všechna tato opatření při přípravě léčivých přípravků vyráběných z lidské krve nebo plazmy nelze riziko přenosu infekce zcela vyloučit. Důrazně se doporučuje zaznamenat při každém podání přípravku HyQvia pacientovi název a číslo šarže přípravku, aby bylo možné zpětně přiřadit k pacientovi číslo použité šarže. Pediatrická populace: Uvedená upozornění a opatření platí jako pro dospělé, tak pro děti. Fertilita, těhotenství a kojení: Bezpečnost léčivého přípravku při podávání těhotným ženám nebyla stanovena v kontrolovaných klinických hodnoceních, a proto by se měl tento přípravek podávat těhotným ženám a kojícím matkám pouze s opatrností. Klinické zkušenosti s imunoglobuliny nenaznačují, že by IG 10% měl negativní vliv na fertilitu.
Hlavní nežádoucí účinky: Nejčastěji hlášené nežádoucí účinky (NÚ) přípravku HyQvia jsou lokální reakce. Nejčastěji hlášenými systémovými NÚ byly bolest hlavy, únava a pyrexie. Většina NÚ byla mírná až středně závažná. Normální lidský imunoglobulin: Příležitostně se mohou objevit nežádoucí účinky, jako je třesavka, bolest hlavy, závratě, horečka, zvracení, alergické reakce, nevolnost, artralgie, nízký krevní tlak a středně závažná bolest dolní poloviny zad. Často se mohou vyskytnout lokální reakce v místech infuze: zduření, bolestivost, erytém, indurace, lokální zahřátí, svědění, zhmoždění a vyrážka. Rekombinantní lidská hyaluronidáza: Nejčastější NÚ uváděné během postmarketingového užívání rekombinantní lidské hyaluronidázy v podobném složení podávané subkutánně za účelem disperze a absorpce subkutánně aplikovaných tekutin nebo léčivých přípravků byly mírné lokální reakce v místě infuze jako např. erytém a bolest. V souvislosti s aplikací velkého objemu subkutánních tekutin byl nejčastěji hlášen otok. NÚ hlášené v klinických studiích s frekvencí velmi časté (≥1/10) byly lokální reakce (celkové) a bolest v místě infuze (včetně diskomfortu, citlivosti, bolesti třísla). Významné interakce: Aplikace imunoglobulinu může na dobu nejméně 6 týdnů a nejvýše 3 měsíců narušit účinnost živých atenuovaných virových vakcín. V případě spalniček může toto narušení účinnosti trvat až 1 rok. Proto je potřeba u pacientů očkovaných vakcínou proti spalničkám zkontrolovat stav protilátek.
Uchovávání: Uchovávejte v chladničce (2°C – 8°C). Chraňte před mrazem. Uchovávejte injekční lahvičky v krabičce, aby byl přípravek chráněn před světlem.
Držitel rozhodnutí o registraci: Baxalta Innovations GmbH, Industriestrasse 67, A1221 Vídeň, Rakousko.
Registrační čísla: 2,5g/25ml EU/1/13/840/001, 5g/50ml EU/1/13/840/002, 10g/100ml EU/1/13/840/003, 20g/200ml EU/1/13/840/004, 30g/300ml EU/1/13/840/005
Poslední revize SPC: 01/2020
Výdej léčivého přípravku je vázán na lékařský předpis.
Léčivý přípravek je hrazen z prostředků veřejného zdravotního pojištění.
Úplné znění SPC naleznete na www.sukl.cz.
Žádáme zdravotnické pracovníky, aby hlásili podezření na nežádoucí účinky na SÚKL nebo společnosti Takeda emailem na drugsafety-cz@takeda.com. Podezření na nežádoucí účinky hlaste také podle národních legislativních požadavků.
Dříve než začnete přípravek předepisovat, seznamte se, prosím, s úplným souhrnem údajů o přípravku (SPC).
▼ Tento léčivý přípravek podléhá dalšímu sledování. To umožní rychlé získání nových informací o bezpečnosti. Žádáme zdravotnické pracovníky, aby hlásili jakákoli podezření na nežádoucí účinky. Podrobnosti o hlášení nežádoucích účinků viz bod 4.8 SPC.
Složení: Jedna injekční lahvička immunoglobulinum humanum normale (IG 10%) a jedna injekční lahvička hyaluronidasum humanum biosyntheticum (rHuPH20). Léčivá látka: Jeden ml obsahuje 100 mg normálního lidského imunoglobulinu (o čistotě alespoň 98 % IgG). Maximální obsah IgA je 140 mikrogramů/ml. Pomocné látky: Rekombinantní lidská hyaluronidáza (rHuPH20) je purifikovaný glykoprotein o 447 aminokyselinách produkovaný buňkami vaječníku čínského křečíka (CHO) technologií rekombinantní DNA. Injekční lahvička normálního lidského imunoglobulinu (IG 10%): Glycin, voda pro injekci. Injekční lahvička rekombinantní lidské hyaluronidázy (rHuPH20): Chlorid sodný, hydrogenfosforečnan sodný, lidský albumin, dinatriumedetát (EDTA), chlorid vápenatý, hydroxid sodný (pro úpravu pH), kyselina chlorovodíková (pro úpravu pH), voda pro injekci.
Indikace: Substituční terapie dospělých, dětí a dospívajících (0–18 let) u syndromů primární imunodeficience s narušenou tvorbou protilátek, u hypogamaglobulinemie a rekurentních bakteriálních infekcí u pacientů s chronickou lymfatickou leukemií (CLL), u nichž selhala profylaktická antibiotika nebo jsou kontraindikována, u hypogamaglobulinemie a rekurentních bakteriálních infekcí u pacientů s mnohočetným myelomem (MM) a u hypogamaglobulinemie u pacientů před a po alogenní transplantaci hematopoetických kmenových buněk (HSCT).
Dávkování a způsob podání: Substituční terapie by měla být zahájena a monitorována lékařem zkušeným v léčbě imunodeficience. Dávka a dávkovací režim závisí na indikaci. Při substituční terapii je dávka u každého pacienta individuální, závisí na konkrétní farmakokinetice a klinické odpovědi. Následující dávkovací režimy jsou pouze orientační. Pacienti dosud neléčení imunoglobuliny: Dávka potřebná k dosažení minimální hladiny 6 g/l v rovnovážném stavu je řádově 0,4–0,8 g/kg/měsíc. Dávkovací interval k udržení hladin v rovnovážném stavu se liší v rozmezí 2–4 týdnů. Kumulativní měsíční dávka IG 10% by měla být rozdělena do jednotýdenních, dvoutýdenních atd. dávek podle naplánovaných léčebných intervalů přípravkem HyQvia. Pacienti dříve léčení imunoglobulinem podávaným intravenózně: Léčivý přípravek by měl být podáván ve stejné dávce a se stejnou četností jako jejich předchozí intravenózní imunoglobulinová léčba. Pokud byli dříve pacienti na 3týdenním dávkovacím režimu, zvýšení intervalu na 4 týdny lze dosáhnout podáváním stejných týdenních ekvivalentů. Pacienti dříve léčení subkutánně podávaným imunoglobulinem: Úvodní dávka přípravku HyQvia je stejná jako u subkutánní léčby, lze ji však přizpůsobit 3 až 4týdennímu intervalu. První infuze přípravku HyQvia by měla být podána jeden týden po poslední léčbě předchozím imunoglobulinem. Pediatrická populace: Dávkování u dětí a dospívajících (0–18 let) se neliší od dávkování u dospělých, neboť dávkování v každé indikaci je dáno tělesnou hmotností a upravuje se podle klinického výsledku výše zmíněného onemocnění. Způsob podání: pouze k subkutánnímu podání, nepodávejte intravenózně. Přípravek HyQvia je tvořen dvěma injekčními lahvičkami. Každá injekční lahvička IG 10% je dodávána s odpovídajícím množstvím rekombinantní lidské hyaluronidázy, viz informace v SPC.
Kontraindikace: HyQvia se nesmí podávat intravenózně ani intramuskulárně. Hypersenzitivita na léčivou látku (IgG) nebo na kteroukoli pomocnou látku. Hypersenzitivita na lidské imunoglobuliny, zejména ve velmi vzácných případech deficitu IgA, kdy má pacient proti IgA protilátky. Známá systémová hypersenzitivita na hyaluronidázu nebo rekombinantní lidskou hyaluronidázu.
Upozornění: Pokud je přípravek HyQvia náhodně aplikován do žíly, může u pacienta vyvolat šok. Používejte doporučené rychlosti infuze. Pacienty je nutné důsledně sledovat v průběhu celé infuze, a to především pacienty začínající s léčbou. Určité nežádoucí účinky se mohou objevovat častěji u pacientů, kteří dostávají normální lidský imunoglobulin poprvé nebo (ve vzácných případech) jej mění nebo pokud uběhla dlouhá doba od předchozí infuze. V případě nežádoucích účinků je nutné buď snížit rychlost podávání infuze, nebo ji úplně zastavit. V případě šoku okamžitě ukončete infuzi a zahajte u pacienta léčbu šoku. Hypersenzitivita na IG 10%: Pravé reakce přecitlivělosti jsou vzácné. Může k nim docházet především u pacientů s protilátkami proti IgA, které je třeba léčit se zvýšenou opatrností. Hypersenzitivita na rekombinantní lidskou hyaluronidázu: Jakékoli podezření na reakci podobnou alergické nebo anafylaktické reakci po podání rekombinantní lidské hyaluronidázy vyžaduje okamžité přerušení infuze a – podle konkrétní potřeby – zahájení standardního léčebného postupu. Imunogenicita rekombinantní lidské hyaluronidázy: U pacientů léčených přípravkem HyQvia v klinických studiích byl hlášen vznik jiných než neutralizačních protilátek proti rekombinantní lidské hyaluronidáze. Tromboembolismus: S použitím imunoglobulinů byly spojeny arteriální a venózní tromboembolické příhody, včetně infarktu myokardu, mozkové příhody, hluboké žilní trombózy a plicní embolie. Před použitím imunoglobulinů musí být pacienti dostatečně hydratováni. U pacientů s preexistujícími rizikovými faktory výskytu tromboembolické příhody je třeba postupovat s opatrností. Hemolytická anémie. Imunoglobulinové přípravky obsahují protilátky proti krevním skupinám (např. A, B, D), které se mohou chovat jako hemolyziny. Syndrom aseptické meningitidy (AMS): Ve spojení s intravenózní a subkutánní imunoglobulinovou léčbou byl hlášen výskyt syndromu aseptické meningitidy. AMS se může častěji objevovat ve spojitosti s vysokou dávkou (2 g/kg) intravenózní imunoglobulinovou léčbou. Důležité informace o některých složkách přípravku HyQvia: Přípravek HyQvia neobsahuje cukry. Složka IG 10% obsahuje stopová množství sodíku. Rekombinantní lidská hyaluronidáza obsahuje 4,03 mg sodíku na ml, s maximální denní dávkou přibližně 120 mg. Interference se sérologickými testy: Po imunoglobulinové infuzi může mít přechodný vzestup různých pasivně přenášených protilátek v krvi pacienta za následek zavádějící pozitivní výsledky sérologických testů. Přenosná agens: Normální lidský imunoglobulin a lidský sérový albumin (stabilizátor rekombinantní lidské hyaluronidázy) se vyrábějí z lidské plazmy. Standardní opatření zabraňující přenosu infekce zahrnují pečlivý výběr dárců, testování jednotlivých odběrů krve a plazmatických poolů na specifické ukazatele infekce a určité výrobní kroky, při nichž jsou inaktivovány nebo odstraněny viry. Přes všechna tato opatření při přípravě léčivých přípravků vyráběných z lidské krve nebo plazmy nelze riziko přenosu infekce zcela vyloučit. Důrazně se doporučuje zaznamenat při každém podání přípravku HyQvia pacientovi název a číslo šarže přípravku, aby bylo možné zpětně přiřadit k pacientovi číslo použité šarže. Pediatrická populace: Uvedená upozornění a opatření platí jako pro dospělé, tak pro děti. Fertilita, těhotenství a kojení: Bezpečnost léčivého přípravku při podávání těhotným ženám nebyla stanovena v kontrolovaných klinických hodnoceních, a proto by se měl tento přípravek podávat těhotným ženám a kojícím matkám pouze s opatrností. Klinické zkušenosti s imunoglobuliny nenaznačují, že by IG 10% měl negativní vliv na fertilitu.
Hlavní nežádoucí účinky: Nejčastěji hlášené nežádoucí účinky (NÚ) přípravku HyQvia jsou lokální reakce. Nejčastěji hlášenými systémovými NÚ byly bolest hlavy, únava a pyrexie. Většina NÚ byla mírná až středně závažná. Normální lidský imunoglobulin: Příležitostně se mohou objevit nežádoucí účinky, jako je třesavka, bolest hlavy, závratě, horečka, zvracení, alergické reakce, nevolnost, artralgie, nízký krevní tlak a středně závažná bolest dolní poloviny zad. Často se mohou vyskytnout lokální reakce v místech infuze: zduření, bolestivost, erytém, indurace, lokální zahřátí, svědění, zhmoždění a vyrážka. Rekombinantní lidská hyaluronidáza: Nejčastější NÚ uváděné během postmarketingového užívání rekombinantní lidské hyaluronidázy v podobném složení podávané subkutánně za účelem disperze a absorpce subkutánně aplikovaných tekutin nebo léčivých přípravků byly mírné lokální reakce v místě infuze jako např. erytém a bolest. V souvislosti s aplikací velkého objemu subkutánních tekutin byl nejčastěji hlášen otok. NÚ hlášené v klinických studiích s frekvencí velmi časté (≥1/10) byly lokální reakce (celkové) a bolest v místě infuze (včetně diskomfortu, citlivosti, bolesti třísla). Významné interakce: Aplikace imunoglobulinu může na dobu nejméně 6 týdnů a nejvýše 3 měsíců narušit účinnost živých atenuovaných virových vakcín. V případě spalniček může toto narušení účinnosti trvat až 1 rok. Proto je potřeba u pacientů očkovaných vakcínou proti spalničkám zkontrolovat stav protilátek.
Uchovávání: Uchovávejte v chladničce (2°C – 8°C). Chraňte před mrazem. Uchovávejte injekční lahvičky v krabičce, aby byl přípravek chráněn před světlem.
Držitel rozhodnutí o registraci: Baxalta Innovations GmbH, Industriestrasse 67, A1221 Vídeň, Rakousko.
Registrační čísla: 2,5g/25ml EU/1/13/840/001, 5g/50ml EU/1/13/840/002, 10g/100ml EU/1/13/840/003, 20g/200ml EU/1/13/840/004, 30g/300ml EU/1/13/840/005
Poslední revize SPC: 01/2020
Výdej léčivého přípravku je vázán na lékařský předpis.
Léčivý přípravek je hrazen z prostředků veřejného zdravotního pojištění.
Úplné znění SPC naleznete na www.sukl.cz.
Žádáme zdravotnické pracovníky, aby hlásili podezření na nežádoucí účinky na SÚKL nebo společnosti Takeda emailem na drugsafety-cz@takeda.com. Podezření na nežádoucí účinky hlaste také podle národních legislativních požadavků.
Cuvitru 200 mg/ml injekční roztok
Zkrácené informace o přípravku
Dříve než začnete přípravek předepisovat, seznamte se, prosím, s úplným Souhrnem údajů o přípravku.
▼ Tento léčivý přípravek podléhá dalšímu sledování. To umožní rychlé získání nových informací o bezpečnosti. Žádáme zdravotnické pracovníky, aby hlásili jakákoli podezření na nežádoucí účinky. Podrobnosti o hlášení nežádoucích účinků viz bod 4.8 SPC.
Složení: Léčivá látka: Jeden ml obsahuje Immunoglobulinum humanum normale 200 mg (o čistotě alespoň 98 % IgG). Maximální obsah IgA je 280 mikrogramů/ml. Pomocné látky: Glycin, voda na injekci.
Indikace: Indikace pro subkutánní podání (SCIg). Substituční terapie u dospělých a dětí a dospívajících (0–18 let) u syndromů primární imunodeficience s narušenou tvorbou protilátek; hypogamaglobulinemie a rekurentních bakteriálních infekcí u pacientů s chronickou lymfatickou leukemií (CLL), u nichž selhala profylaktická antibiotika nebo jsou kontraindikována; hypogamaglobulinemie a rekurentních bakteriálních infekcí u pacientů s mnohočetným myelomem (MM); hypogamaglobulinemie u pacientů před a po alogenní transplantaci hematopoetických kmenových buněk (hematopoietic stem cell transplantation, HSCT).
Dávkování a způsob podání: Substituční terapie má být zahájena a monitorována lékařem se zkušeností s léčbou imunodeficience. Dávka a dávkovací režim závisí na indikaci. Přípravek je určen k subkutánnímu podání. Při substituční terapii je nutné dávku u každého pacienta stanovit individuálně v závislosti na konkrétní farmakokinetice a klinické odpovědi. Dávkovací režim má dosáhnout minimální hladiny IgG (měřené před další infuzí) alespoň 5 až 6 g/l a cílová hladina se má nacházet v referenčním intervalu sérové hladiny IgG pro daný věk. Dávkování u dětí a dospívajících (0–18 let) se neliší od dospělých, neboť dávkování pro každou indikaci se stanoví na základě tělesné hmotnosti a upraví se podle klinického výsledku výše zmíněných indikací.
Kontraindikace: Hypersenzitivita na léčivou látku nebo na kteroukoli pomocnou látku. Závažný deficit IgA a hypersenzitivita na léčbu lidskými imunoglobuliny v anamnéze. Cuvitru se nesmí podávat intravaskulárně nebo intramuskulárně.
Upozornění: Pacienti s protilátkami proti IgA, u nichž zůstává léčba subkutánně podávanými přípravky IgG jedinou možností, mají být léčeni přípravkem Cuvitru pouze pod pečlivým lékařským dohledem. Normální lidský imunoglobulin může vzácně vyvolat pokles krevního tlaku s anafylaktickou reakcí, a to dokonce i u pacientů, kteří předchozí léčbu normálním lidským imunoglobulinem tolerovali. S používáním imunoglobulinu byly spojeny arteriální a venózní tromboembolické příhody. U pacientů s preexistujícími rizikovými faktory výskytu trombotické příhody je třeba postupovat s opatrností. U pacientů léčených imunoglobuliny byly hlášeny závažné renální nežádoucí účinky, zejména v případě přípravků obsahujících sacharózu (Cuvitru neobsahuje sacharózu). Ve spojení s imunoglobulinovou léčbou byl hlášen výskyt AMS. Cuvitru obsahuje protilátky proti krevním skupinám, které mohou působit jako hemolyziny a vyvolat in vivo obalení trombocytů imunoglobulinem. To může vést k pozitivnímu přímému antiglobulinovému testu a vzácně k hemolýze. Po imunoglobulinové injekci může mít přechodný vzestup různých pasivně přenášených protilátek v krvi pacienta za následek zavádějící pozitivní výsledky sérologických testů. Přes všechna standardní opatření zabraňující přenosu infekce v souvislosti s používáním léčivých přípravků vyrobených z lidské krve nebo plazmy nelze při jejich podávání riziko přenosu infekce zcela vyloučit. Toto se vztahuje také na neznámé a nově vznikající viry a jiné typy patogenů. Důrazně se doporučuje zaznamenat název a číslo šarže přípravku, aby bylo možné zpětně přiřadit k pacientovi číslo použité šarže.
Interakce: Aplikace imunoglobulinu může na dobu nejméně 6 týdnů a nejvýše 3 měsíců snížit účinnost živých atenuovaných virových vakcín, jako jsou spalničky, rubeola, příušnice a varicella. Po podání přípravku Cuvitru je třeba vyčkat 3 měsíce před vakcinací živou atenuovanou vakcínou. V případě spalniček může toto narušení účinnosti trvat až 1 rok. Proto je potřeba u pacientů očkovaných vakcínou proti spalničkám zkontrolovat stav protilátek.
Fertilita, těhotenství a kojení: Cuvitru se má podávat těhotným a kojícím ženám pouze s opatrností. Klinické zkušenosti s imunoglobuliny naznačují, že není třeba očekávat žádné škodlivé účinky na fertilitu.
Řízení: Schopnost řídit a obsluhovat stroje může být ovlivněna některými nežádoucími účinky spojenými s přípravkem Cuvitru. Pacienti, kteří během léčby některé nežádoucí účinky prodělají, mají počkat, až odezní, než začnou řídit nebo obsluhovat stroje.
Nežádoucí účinky: Příležitostně se mohou objevit nežádoucí účinky, jako je třesavka, bolest hlavy, závrať, horečka, zvracení, alergické reakce, nauzea, artralgie, nízký krevní tlak a středně závažná bolest dolní části zad. Vzácně může normální lidský imunoglobulin způsobit náhlý pokles krevního tlaku a v izolovaných případech anafylaktický šok, a to i u pacientů, kteří při předchozí aplikaci přípravku nejevili žádné známky hypersenzitivity. Lokální reakce v místě infuze: často se může vyskytnout zduření, bolestivost, erytém, indurace, lokální zahřátí, lokální bolest, svědění, vznik modřin a vyrážka. Velmi časté nežádoucí účinky (četnost ≥ 1/10): Bolest hlavy, průjem, nauzea, lokální reakce, erytém v místě infuze, bolest v místě injekce, únava. Časté nežádoucí účinky (četnost ≥ 1/100 až < 1/10): Závrať, migréna, somnolence, hypotenze, bolest břicha, pruritus, kopřivka, myalgie, zduření v místě infuze, pruritus v místě injekce, kopřivka v místě infuze, podlitina v místě infuze a bolest.
Předávkování: Následky předávkování nejsou známy.
Uchovávání: Neuchovávejte při teplotě nad 25 °C. Chraňte před mrazem. Uchovávejte injekční lahvičku v krabičce, aby byl přípravek chráněn před světlem.
Držitel rozhodnutí o registraci: Baxalta Innovations GmbH, Industriestrasse 67, A‑1221 Vídeň, Rakousko.
Registrační čísla: 59/646/15-C
Poslední revize SPC: 15.5.2020
Výdej léčivého přípravku je vázán na lékařský předpis
Léčivý přípravek je hrazen z prostředků veřejného zdravotního pojištění.
Úplné znění SPC naleznete na www.sukl.cz.
Žádáme zdravotnické pracovníky, aby hlásili podezření na nežádoucí účinky na SÚKL nebo společnosti Takeda emailem na drugsafety-cz@takeda.com. Podezření na nežádoucí účinky hlaste také podle národních legislativních požadavků.
Dříve než začnete přípravek předepisovat, seznamte se, prosím, s úplným Souhrnem údajů o přípravku.
▼ Tento léčivý přípravek podléhá dalšímu sledování. To umožní rychlé získání nových informací o bezpečnosti. Žádáme zdravotnické pracovníky, aby hlásili jakákoli podezření na nežádoucí účinky. Podrobnosti o hlášení nežádoucích účinků viz bod 4.8 SPC.
Složení: Léčivá látka: Jeden ml obsahuje Immunoglobulinum humanum normale 200 mg (o čistotě alespoň 98 % IgG). Maximální obsah IgA je 280 mikrogramů/ml. Pomocné látky: Glycin, voda na injekci.
Indikace: Indikace pro subkutánní podání (SCIg). Substituční terapie u dospělých a dětí a dospívajících (0–18 let) u syndromů primární imunodeficience s narušenou tvorbou protilátek; hypogamaglobulinemie a rekurentních bakteriálních infekcí u pacientů s chronickou lymfatickou leukemií (CLL), u nichž selhala profylaktická antibiotika nebo jsou kontraindikována; hypogamaglobulinemie a rekurentních bakteriálních infekcí u pacientů s mnohočetným myelomem (MM); hypogamaglobulinemie u pacientů před a po alogenní transplantaci hematopoetických kmenových buněk (hematopoietic stem cell transplantation, HSCT).
Dávkování a způsob podání: Substituční terapie má být zahájena a monitorována lékařem se zkušeností s léčbou imunodeficience. Dávka a dávkovací režim závisí na indikaci. Přípravek je určen k subkutánnímu podání. Při substituční terapii je nutné dávku u každého pacienta stanovit individuálně v závislosti na konkrétní farmakokinetice a klinické odpovědi. Dávkovací režim má dosáhnout minimální hladiny IgG (měřené před další infuzí) alespoň 5 až 6 g/l a cílová hladina se má nacházet v referenčním intervalu sérové hladiny IgG pro daný věk. Dávkování u dětí a dospívajících (0–18 let) se neliší od dospělých, neboť dávkování pro každou indikaci se stanoví na základě tělesné hmotnosti a upraví se podle klinického výsledku výše zmíněných indikací.
Kontraindikace: Hypersenzitivita na léčivou látku nebo na kteroukoli pomocnou látku. Závažný deficit IgA a hypersenzitivita na léčbu lidskými imunoglobuliny v anamnéze. Cuvitru se nesmí podávat intravaskulárně nebo intramuskulárně.
Upozornění: Pacienti s protilátkami proti IgA, u nichž zůstává léčba subkutánně podávanými přípravky IgG jedinou možností, mají být léčeni přípravkem Cuvitru pouze pod pečlivým lékařským dohledem. Normální lidský imunoglobulin může vzácně vyvolat pokles krevního tlaku s anafylaktickou reakcí, a to dokonce i u pacientů, kteří předchozí léčbu normálním lidským imunoglobulinem tolerovali. S používáním imunoglobulinu byly spojeny arteriální a venózní tromboembolické příhody. U pacientů s preexistujícími rizikovými faktory výskytu trombotické příhody je třeba postupovat s opatrností. U pacientů léčených imunoglobuliny byly hlášeny závažné renální nežádoucí účinky, zejména v případě přípravků obsahujících sacharózu (Cuvitru neobsahuje sacharózu). Ve spojení s imunoglobulinovou léčbou byl hlášen výskyt AMS. Cuvitru obsahuje protilátky proti krevním skupinám, které mohou působit jako hemolyziny a vyvolat in vivo obalení trombocytů imunoglobulinem. To může vést k pozitivnímu přímému antiglobulinovému testu a vzácně k hemolýze. Po imunoglobulinové injekci může mít přechodný vzestup různých pasivně přenášených protilátek v krvi pacienta za následek zavádějící pozitivní výsledky sérologických testů. Přes všechna standardní opatření zabraňující přenosu infekce v souvislosti s používáním léčivých přípravků vyrobených z lidské krve nebo plazmy nelze při jejich podávání riziko přenosu infekce zcela vyloučit. Toto se vztahuje také na neznámé a nově vznikající viry a jiné typy patogenů. Důrazně se doporučuje zaznamenat název a číslo šarže přípravku, aby bylo možné zpětně přiřadit k pacientovi číslo použité šarže.
Interakce: Aplikace imunoglobulinu může na dobu nejméně 6 týdnů a nejvýše 3 měsíců snížit účinnost živých atenuovaných virových vakcín, jako jsou spalničky, rubeola, příušnice a varicella. Po podání přípravku Cuvitru je třeba vyčkat 3 měsíce před vakcinací živou atenuovanou vakcínou. V případě spalniček může toto narušení účinnosti trvat až 1 rok. Proto je potřeba u pacientů očkovaných vakcínou proti spalničkám zkontrolovat stav protilátek.
Fertilita, těhotenství a kojení: Cuvitru se má podávat těhotným a kojícím ženám pouze s opatrností. Klinické zkušenosti s imunoglobuliny naznačují, že není třeba očekávat žádné škodlivé účinky na fertilitu.
Řízení: Schopnost řídit a obsluhovat stroje může být ovlivněna některými nežádoucími účinky spojenými s přípravkem Cuvitru. Pacienti, kteří během léčby některé nežádoucí účinky prodělají, mají počkat, až odezní, než začnou řídit nebo obsluhovat stroje.
Nežádoucí účinky: Příležitostně se mohou objevit nežádoucí účinky, jako je třesavka, bolest hlavy, závrať, horečka, zvracení, alergické reakce, nauzea, artralgie, nízký krevní tlak a středně závažná bolest dolní části zad. Vzácně může normální lidský imunoglobulin způsobit náhlý pokles krevního tlaku a v izolovaných případech anafylaktický šok, a to i u pacientů, kteří při předchozí aplikaci přípravku nejevili žádné známky hypersenzitivity. Lokální reakce v místě infuze: často se může vyskytnout zduření, bolestivost, erytém, indurace, lokální zahřátí, lokální bolest, svědění, vznik modřin a vyrážka. Velmi časté nežádoucí účinky (četnost ≥ 1/10): Bolest hlavy, průjem, nauzea, lokální reakce, erytém v místě infuze, bolest v místě injekce, únava. Časté nežádoucí účinky (četnost ≥ 1/100 až < 1/10): Závrať, migréna, somnolence, hypotenze, bolest břicha, pruritus, kopřivka, myalgie, zduření v místě infuze, pruritus v místě injekce, kopřivka v místě infuze, podlitina v místě infuze a bolest.
Předávkování: Následky předávkování nejsou známy.
Uchovávání: Neuchovávejte při teplotě nad 25 °C. Chraňte před mrazem. Uchovávejte injekční lahvičku v krabičce, aby byl přípravek chráněn před světlem.
Držitel rozhodnutí o registraci: Baxalta Innovations GmbH, Industriestrasse 67, A‑1221 Vídeň, Rakousko.
Registrační čísla: 59/646/15-C
Poslední revize SPC: 15.5.2020
Výdej léčivého přípravku je vázán na lékařský předpis
Léčivý přípravek je hrazen z prostředků veřejného zdravotního pojištění.
Úplné znění SPC naleznete na www.sukl.cz.
Žádáme zdravotnické pracovníky, aby hlásili podezření na nežádoucí účinky na SÚKL nebo společnosti Takeda emailem na drugsafety-cz@takeda.com. Podezření na nežádoucí účinky hlaste také podle národních legislativních požadavků.
KIOVIG 100 mg/ml, infuzní roztok
Zkrácené informace o přípravku
Složení: Léčivá látka: Immunoglobulinum humanum normale (IVIg) 100 mg/ml (čistota nejméně 98 % IgG). Jedna injekční lahvička 10 ml (25 ml, 50 ml, 100 ml, 200 ml, 300 ml, resp.) obsahuje: immunoglobulinum humanum normale 1 g (2,5 g, 5 g, 10 g, 20 g, 30 g, resp.). Maximální obsah imunoglobulinu A (IgA): 140 mikrogramů/ml. Pomocné látky: glycin, voda na injekci.
Indikace: Substituční léčba u syndromů primárního imunodeficitu (PID) s poruchou tvorby protilátek; sekundárních imunodeficitů (SID) u pacientů trpících závažnými nebo opakovanými infekcemi, neúčinnou antimikrobiální léčbou, a buď s prokázaným selháním specifické protilátky (PSAF)#, nebo s hladinou IgG v séru < 4 g/l. #PSAF = neschopnost dosáhnout alespoň 2násobného zvýšení v titru protilátky IgG u pneumokokových vakcín s polysacharidovým nebo polypeptidovým antigenem; Imunomodulace u primární imunitní trombocytopenie u pacientů s vysokým rizikem krvácení nebo před operací za účelem korekce počtu krevních destiček, Guillain-Barrého syndromu, Kawasakiho choroby (v kombinaci s kyselinou acetylsalicylovou), chronické zánětlivé demyelinizační polyradikuloneuropatie (CIDP), multifokální motorické neuropatie (MMN).
Dávkování a způsob podání:* Dávka a dávkovací režim závisí na indikaci. Dávkování v rámci substituční léčby je u každého pacienta individuální a závisí na farmakokinetické a klinické odezvě. U pacientů s podváhou nebo nadváhou může být nutné upravit dávku dle tělesné hmotnosti. Následující režimy dávkování jsou uváděny jako doporučení: Substituční léčba u primárního imunodeficitu: úvodní dávka 0,4–0,8 g/kg, poté 0,2-0,8 g/kg každé 3-4 týdny pro zajištění minimální hladiny IgG alespoň 5-6 g/l. Sekundární imunodeficity: 0,2–0,4 g/kg každé 3-4 týdny pro zajištění minimální hladiny IgG alespoň 5-6 g/l. Primární imunitní trombocytopenie: 0,8–1 g/kg první den, tuto dávku lze opakovat jednou za tři dny, nebo 0,4 g/kg/den po dobu 2-5 dnů. Guillain-Barrého syndrom: 0,4 g/kg/den po dobu 5 dnů (možné opakované dávkování v případě relapsu). Kawasakiho choroba. 2,0 g/kg v jedné dávce v kombinaci s kyselinou acetylsalicylovou. Chronická zánětlivá demyelinizační polyradikuloneuropatie (CIDP): Úvodní dávka 2 g/kg rozdělená do 2-5 po sobě následujících dnů. Udržovací dávky 1 g/kg během 1-2 po sobě následujících dnů každé 3 týdny. Po každém cyklu je nutno vyhodnotit účinek léčby: pokud po 6 měsících není pozorován žádný léčebný účinek, léčba má být ukončena. Pokud je léčba účinná, o dlouhodobé léčbě rozhodne lékař dle svého uvážení na základě odpovědi pacienta na léčbu a odpovědi na její udržování. Může být nutné upravit dávkování a intervaly dle individuálního průběhu onemocnění. Multifokální motorická neuropatie: Úvodní dávka: 2 g/kg po dobu 2–5 po sobě jdoucích dnů. Udržovací dávka: 1 g/kg každé 2 až 4 týdny nebo 2 g/kg každých 4 až 8 týdnů po dobu 2-5 dnů. Po každém cyklu je nutno vyhodnotit účinek léčby: pokud po 6 měsících není pozorován žádný léčebný účinek, léčba má být ukončena. Pokud je léčba účinná, o dlouhodobé léčbě rozhodne lékař dle svého uvážení na základě odpovědi pacienta na léčbu a odpovědi na její udržování. Může být nutné upravit dávkování a intervaly dle individuálního průběhu onemocnění. Způsob podání: K intravenóznímu podání. Normální lidský imunoglobulin má být podáván intravenózně počáteční rychlostí 0,5 ml/kg/h po dobu 30 minut. Je-li dobře snášen, může být rychlost podání postupně zvýšena na maximum 6 ml/kg/h po dobu 30 minut. Klinické zkušenosti u omezeného počtu pacientů rovněž ukazují, že dospělí pacienti s PID mohou tolerovat rychlost podání až 8 ml/kg/h. Je-li zapotřebí naředění před infuzí, může být KIOVIG ředěn 5% roztokem glukózy na konečnou koncentraci 50 mg/ml.
Kontraindikace: Hypersenzitivita na léčivou látku nebo na kteroukoli pomocnou látku. Hypersenzitivita na lidské imunoglobuliny, zvláště u pacientů s protilátkami proti IgA. Pacienti se selektivním deficitem IgA, u kterých se vytvořily protilátky na IgA, protože podání přípravku s obsahem IgA může způsobit anafylaxi.
Upozornění: Reakce na infuzi: Některé závažné nežádoucí účinky (např. bolest hlavy, zrudnutí, třesavka, myalgie, sípání, tachykardie, bolest v dolní části zad, nauzea a hypotenze) mohou souviset s rychlostí infuze. Doporučenou rychlost infuze je nutno pečlivě dodržovat. Hypersenzitivní reakce jsou vzácné. Anafylaxe se může rozvinout u pacientů s nezjistitelným IgA, kteří mají protilátky proti IgA, a u pacientů, kteří tolerovali předchozí léčbu lidským normálním imunoglobulinem. V případě šoku je třeba použít běžný lékařský postup pro léčby šoku. U pacientů podstupujících léčbu IVIg byly rovněž hlášeny případy akutního renálního selhání. Mezi tyto nežádoucí účinky patří akutní renální selhání, akutní tubulární nekróza, proximální tubulární nefropatie a osmotická nefróza. Před infuzí IVIg je nutno zhodnotit renální parametry, zejména u pacientů, u nichž se má za to, že mají potenciálně zvýšené riziko rozvoje akutního renálního selhání, a zhodnocení ve vhodných intervalech opakovat. U pacientů ohrožených akutním renálním selháním je nutno podávat IVIg s minimální možnou rychlostí infuze a velikostí dávky. V případě poruchy funkce ledvin je třeba zvážit přerušení léčby IVIg. U pacientů dostávajících IVIg byly hlášeny případy akutního nekardiogenního pulmonálního edému (akutní plicní poranění v souvislosti s transfuzí, TRALI) u pacientů, kterým byl podáván IVIg (včetně přípravku KIOVIG). TRALI se vyznačuje závažnou hypoxií, dyspnoe, tachypnoe, cyanózou, horečkou a hypotenzí. Příznaky TRALI se obvykle rozvinou během nebo do 6 hodin po transfuzi, často do 1–2 hodin. Příjemci IVIg proto musí být monitorování a v případě nežádoucích plicních reakcí je nutno infuzi IVIg ihned zastavit. TRALI je stav, který potenciálně ohrožuje život a vyžaduje okamžitou léčbu na JIP. V souvislosti s léčbou IVIg byl hlášen výskyt syndromu aseptické meningitidy. Pacienti vykazující tyto známky a příznaky mají být důkladně neurologicky vyšetřeni, včetně studií mozkomíšního moku, aby se vyloučily jiné příčiny meningitidy. Přerušení léčby IVIg mělo během několika dní za důsledek vymizení příznaků AMS bez dalších následků. Sekundárně k léčbě IVIg se může rozvinout hemolytická anémie. Po léčbě IVIg bylo hlášeno přechodné snížení počtu neutrofilů a/nebo epizody neutropenie, někdy závažné. Toto obvykle nastává během několika hodin nebo dnů po podání IVIg a odezní spontánně během 7 až 14 dnů. Po aplikaci imunoglobulinu může v krvi pacienta dojít k přechodnému vzestupu pasivně přenesených protilátek, a tím ke vzniku zavádějících pozitivních výsledků u sérologických testů. Podávání přípravku KIOVIG může vést k falešně pozitivním výsledkům analýz k diagnostice plísňových infekcí, které závisí na detekci beta D glukanů. Tento stav může přetrvávat týdny po infuzi přípravku. Přes všechna opatření při přípravě léků vyráběných z lidské krve nebo plazmy nelze možnost přenosu infekčních agens zcela vyloučit. Pediatričtí pacienti mohou být citlivější na objemové přetížení.
Významné interakce: Podávání imunoglobulinů může na dobu minimálně 6 týdnů a maximálně 3 měsíců snížit účinnost živých atenuovaných virových vakcín. Mezi podáním tohoto přípravku a vakcinací živou atenuovanou virovou vakcínou by měla uplynout doba 3 měsíců. U spalniček může toto snížení účinnosti trvat až 1 rok. Vyhněte se podání kličkových diuretik.
Hlavní nežádoucí účinky: Souhrn bezpečnostního profilu: Příležitostně se mohou objevit nežádoucí účinky jako je třesavka, bolest hlavy, závrať, horečka, zvracení, alergické reakce, nevolnost, artralgie, pokles krevního tlaku a mírná bolest v dolní části zad. Vzácně mohou normální lidské imunoglobuliny způsobit náhlý pokles krevního tlaku a v ojedinělých případech anafylaktický šok, a to i v případě, že se při předchozí aplikaci přecitlivělost nevyskytla. Po podání normálního lidského imunoglobulinu byly pozorovány případy reverzibilní aseptické meningitidy, vzácné případy přechodných kožních reakcí (včetně kožního lupus erythematosus – četnost neznámá), případy reverzibilní hemolytické reakce, a to zejména u pacientů s krevními skupinami A, B a AB. Po vysokých dávkách léčby IVIg se ve vzácných případech může rozvinout hemolytická anémie vyžadující transfuzi. Byl pozorován vzestup hladiny sérového kreatininu a/nebo akutní selhání ledvin. Velmi vzácně: tromboembolické reakce jako jsou infarkt myokardu, mozková příhoda, plicní embolie a hluboké žilní trombózy. Případy akutního plicního poranění v souvislosti s transfuzí (TRALI). Nežádoucí účinky hlášené během klinických studií: Velmi časté: bolest hlavy, hypertenze, nauzea, vyrážka, lokální reakce (např. bolest/otok/reakce/svědění v místě infuze), horečka a únava. Časté: bronchitida, nasofaryngitida, anémie, lymfadenopatie, snížená chuť k jídlu, nespavost, úzkost, závrať, migréna, parestezie, hypestezie, konjunktivitida, tachykardie, návaly, kašel, vodnatý výtok z nosu, astma, nazální kongesce, orofaryngeální bolest, dyspnoe, průjem, zvracení, bolest břicha, dyspepsie, kontuze, svědění, kopřivka, dermatitida, erytém, bolest zad, artralgie, bolest končetin, myalgie, svalové křeče, svalová slabost, třesavka, edém, onemocnění podobné chřipce, hrudní diskomfort, bolest na hrudi, astenie, malátnost, ztuhlost. Nežádoucí účinky hlášené po uvedení na trh: Frekvence není známa: hemolýza, anafylaktický šok, tranzitorní ischemická ataka, cévní mozková příhoda, infarkt myokardu, hypotenze, hluboká žilní trombóza, plicní embolie, plicní edém, pozitivní přímý Coombsův test, snížená saturace kyslíkem, akutní plicní poranění v souvislosti s transfuzí.
Uchovávání: Uchovávejte při teplotě do 25 °C. Chraňte před mrazem. Uchovávejte vnitřní obal v krabičce, aby byl přípravek chráněn před světlem. Držitel rozhodnutí o registraci: Takeda Manufacturing Austria AG, Industriestrasse 67, A-1221 Vídeň, Rakousko. Registrační čísla: EU/1/05/329/001-006. Poslední revize SPC: 05/2020.
*Všimněte si, prosím, změn v informacích o léčivém přípravku
Dříve než začnete přípravek předepisovat, seznamte se, prosím, s úplným souhrnem údajů o přípravku (SPC). Výdej léčivého přípravku je vázán na lékařský předpis. Léčivý přípravek je hrazen z prostředků veřejného zdravotního pojištění. Úplné znění SPC naleznete na www.sukl.cz.
Žádáme zdravotnické pracovníky, aby hlásili podezření na nežádoucí účinky na SÚKL nebo společnosti Takeda emailem na drugsafety-cz@takeda.com.
Složení: Léčivá látka: Immunoglobulinum humanum normale (IVIg) 100 mg/ml (čistota nejméně 98 % IgG). Jedna injekční lahvička 10 ml (25 ml, 50 ml, 100 ml, 200 ml, 300 ml, resp.) obsahuje: immunoglobulinum humanum normale 1 g (2,5 g, 5 g, 10 g, 20 g, 30 g, resp.). Maximální obsah imunoglobulinu A (IgA): 140 mikrogramů/ml. Pomocné látky: glycin, voda na injekci.
Indikace: Substituční léčba u syndromů primárního imunodeficitu (PID) s poruchou tvorby protilátek; sekundárních imunodeficitů (SID) u pacientů trpících závažnými nebo opakovanými infekcemi, neúčinnou antimikrobiální léčbou, a buď s prokázaným selháním specifické protilátky (PSAF)#, nebo s hladinou IgG v séru < 4 g/l. #PSAF = neschopnost dosáhnout alespoň 2násobného zvýšení v titru protilátky IgG u pneumokokových vakcín s polysacharidovým nebo polypeptidovým antigenem; Imunomodulace u primární imunitní trombocytopenie u pacientů s vysokým rizikem krvácení nebo před operací za účelem korekce počtu krevních destiček, Guillain-Barrého syndromu, Kawasakiho choroby (v kombinaci s kyselinou acetylsalicylovou), chronické zánětlivé demyelinizační polyradikuloneuropatie (CIDP), multifokální motorické neuropatie (MMN).
Dávkování a způsob podání:* Dávka a dávkovací režim závisí na indikaci. Dávkování v rámci substituční léčby je u každého pacienta individuální a závisí na farmakokinetické a klinické odezvě. U pacientů s podváhou nebo nadváhou může být nutné upravit dávku dle tělesné hmotnosti. Následující režimy dávkování jsou uváděny jako doporučení: Substituční léčba u primárního imunodeficitu: úvodní dávka 0,4–0,8 g/kg, poté 0,2-0,8 g/kg každé 3-4 týdny pro zajištění minimální hladiny IgG alespoň 5-6 g/l. Sekundární imunodeficity: 0,2–0,4 g/kg každé 3-4 týdny pro zajištění minimální hladiny IgG alespoň 5-6 g/l. Primární imunitní trombocytopenie: 0,8–1 g/kg první den, tuto dávku lze opakovat jednou za tři dny, nebo 0,4 g/kg/den po dobu 2-5 dnů. Guillain-Barrého syndrom: 0,4 g/kg/den po dobu 5 dnů (možné opakované dávkování v případě relapsu). Kawasakiho choroba. 2,0 g/kg v jedné dávce v kombinaci s kyselinou acetylsalicylovou. Chronická zánětlivá demyelinizační polyradikuloneuropatie (CIDP): Úvodní dávka 2 g/kg rozdělená do 2-5 po sobě následujících dnů. Udržovací dávky 1 g/kg během 1-2 po sobě následujících dnů každé 3 týdny. Po každém cyklu je nutno vyhodnotit účinek léčby: pokud po 6 měsících není pozorován žádný léčebný účinek, léčba má být ukončena. Pokud je léčba účinná, o dlouhodobé léčbě rozhodne lékař dle svého uvážení na základě odpovědi pacienta na léčbu a odpovědi na její udržování. Může být nutné upravit dávkování a intervaly dle individuálního průběhu onemocnění. Multifokální motorická neuropatie: Úvodní dávka: 2 g/kg po dobu 2–5 po sobě jdoucích dnů. Udržovací dávka: 1 g/kg každé 2 až 4 týdny nebo 2 g/kg každých 4 až 8 týdnů po dobu 2-5 dnů. Po každém cyklu je nutno vyhodnotit účinek léčby: pokud po 6 měsících není pozorován žádný léčebný účinek, léčba má být ukončena. Pokud je léčba účinná, o dlouhodobé léčbě rozhodne lékař dle svého uvážení na základě odpovědi pacienta na léčbu a odpovědi na její udržování. Může být nutné upravit dávkování a intervaly dle individuálního průběhu onemocnění. Způsob podání: K intravenóznímu podání. Normální lidský imunoglobulin má být podáván intravenózně počáteční rychlostí 0,5 ml/kg/h po dobu 30 minut. Je-li dobře snášen, může být rychlost podání postupně zvýšena na maximum 6 ml/kg/h po dobu 30 minut. Klinické zkušenosti u omezeného počtu pacientů rovněž ukazují, že dospělí pacienti s PID mohou tolerovat rychlost podání až 8 ml/kg/h. Je-li zapotřebí naředění před infuzí, může být KIOVIG ředěn 5% roztokem glukózy na konečnou koncentraci 50 mg/ml.
Kontraindikace: Hypersenzitivita na léčivou látku nebo na kteroukoli pomocnou látku. Hypersenzitivita na lidské imunoglobuliny, zvláště u pacientů s protilátkami proti IgA. Pacienti se selektivním deficitem IgA, u kterých se vytvořily protilátky na IgA, protože podání přípravku s obsahem IgA může způsobit anafylaxi.
Upozornění: Reakce na infuzi: Některé závažné nežádoucí účinky (např. bolest hlavy, zrudnutí, třesavka, myalgie, sípání, tachykardie, bolest v dolní části zad, nauzea a hypotenze) mohou souviset s rychlostí infuze. Doporučenou rychlost infuze je nutno pečlivě dodržovat. Hypersenzitivní reakce jsou vzácné. Anafylaxe se může rozvinout u pacientů s nezjistitelným IgA, kteří mají protilátky proti IgA, a u pacientů, kteří tolerovali předchozí léčbu lidským normálním imunoglobulinem. V případě šoku je třeba použít běžný lékařský postup pro léčby šoku. U pacientů podstupujících léčbu IVIg byly rovněž hlášeny případy akutního renálního selhání. Mezi tyto nežádoucí účinky patří akutní renální selhání, akutní tubulární nekróza, proximální tubulární nefropatie a osmotická nefróza. Před infuzí IVIg je nutno zhodnotit renální parametry, zejména u pacientů, u nichž se má za to, že mají potenciálně zvýšené riziko rozvoje akutního renálního selhání, a zhodnocení ve vhodných intervalech opakovat. U pacientů ohrožených akutním renálním selháním je nutno podávat IVIg s minimální možnou rychlostí infuze a velikostí dávky. V případě poruchy funkce ledvin je třeba zvážit přerušení léčby IVIg. U pacientů dostávajících IVIg byly hlášeny případy akutního nekardiogenního pulmonálního edému (akutní plicní poranění v souvislosti s transfuzí, TRALI) u pacientů, kterým byl podáván IVIg (včetně přípravku KIOVIG). TRALI se vyznačuje závažnou hypoxií, dyspnoe, tachypnoe, cyanózou, horečkou a hypotenzí. Příznaky TRALI se obvykle rozvinou během nebo do 6 hodin po transfuzi, často do 1–2 hodin. Příjemci IVIg proto musí být monitorování a v případě nežádoucích plicních reakcí je nutno infuzi IVIg ihned zastavit. TRALI je stav, který potenciálně ohrožuje život a vyžaduje okamžitou léčbu na JIP. V souvislosti s léčbou IVIg byl hlášen výskyt syndromu aseptické meningitidy. Pacienti vykazující tyto známky a příznaky mají být důkladně neurologicky vyšetřeni, včetně studií mozkomíšního moku, aby se vyloučily jiné příčiny meningitidy. Přerušení léčby IVIg mělo během několika dní za důsledek vymizení příznaků AMS bez dalších následků. Sekundárně k léčbě IVIg se může rozvinout hemolytická anémie. Po léčbě IVIg bylo hlášeno přechodné snížení počtu neutrofilů a/nebo epizody neutropenie, někdy závažné. Toto obvykle nastává během několika hodin nebo dnů po podání IVIg a odezní spontánně během 7 až 14 dnů. Po aplikaci imunoglobulinu může v krvi pacienta dojít k přechodnému vzestupu pasivně přenesených protilátek, a tím ke vzniku zavádějících pozitivních výsledků u sérologických testů. Podávání přípravku KIOVIG může vést k falešně pozitivním výsledkům analýz k diagnostice plísňových infekcí, které závisí na detekci beta D glukanů. Tento stav může přetrvávat týdny po infuzi přípravku. Přes všechna opatření při přípravě léků vyráběných z lidské krve nebo plazmy nelze možnost přenosu infekčních agens zcela vyloučit. Pediatričtí pacienti mohou být citlivější na objemové přetížení.
Významné interakce: Podávání imunoglobulinů může na dobu minimálně 6 týdnů a maximálně 3 měsíců snížit účinnost živých atenuovaných virových vakcín. Mezi podáním tohoto přípravku a vakcinací živou atenuovanou virovou vakcínou by měla uplynout doba 3 měsíců. U spalniček může toto snížení účinnosti trvat až 1 rok. Vyhněte se podání kličkových diuretik.
Hlavní nežádoucí účinky: Souhrn bezpečnostního profilu: Příležitostně se mohou objevit nežádoucí účinky jako je třesavka, bolest hlavy, závrať, horečka, zvracení, alergické reakce, nevolnost, artralgie, pokles krevního tlaku a mírná bolest v dolní části zad. Vzácně mohou normální lidské imunoglobuliny způsobit náhlý pokles krevního tlaku a v ojedinělých případech anafylaktický šok, a to i v případě, že se při předchozí aplikaci přecitlivělost nevyskytla. Po podání normálního lidského imunoglobulinu byly pozorovány případy reverzibilní aseptické meningitidy, vzácné případy přechodných kožních reakcí (včetně kožního lupus erythematosus – četnost neznámá), případy reverzibilní hemolytické reakce, a to zejména u pacientů s krevními skupinami A, B a AB. Po vysokých dávkách léčby IVIg se ve vzácných případech může rozvinout hemolytická anémie vyžadující transfuzi. Byl pozorován vzestup hladiny sérového kreatininu a/nebo akutní selhání ledvin. Velmi vzácně: tromboembolické reakce jako jsou infarkt myokardu, mozková příhoda, plicní embolie a hluboké žilní trombózy. Případy akutního plicního poranění v souvislosti s transfuzí (TRALI). Nežádoucí účinky hlášené během klinických studií: Velmi časté: bolest hlavy, hypertenze, nauzea, vyrážka, lokální reakce (např. bolest/otok/reakce/svědění v místě infuze), horečka a únava. Časté: bronchitida, nasofaryngitida, anémie, lymfadenopatie, snížená chuť k jídlu, nespavost, úzkost, závrať, migréna, parestezie, hypestezie, konjunktivitida, tachykardie, návaly, kašel, vodnatý výtok z nosu, astma, nazální kongesce, orofaryngeální bolest, dyspnoe, průjem, zvracení, bolest břicha, dyspepsie, kontuze, svědění, kopřivka, dermatitida, erytém, bolest zad, artralgie, bolest končetin, myalgie, svalové křeče, svalová slabost, třesavka, edém, onemocnění podobné chřipce, hrudní diskomfort, bolest na hrudi, astenie, malátnost, ztuhlost. Nežádoucí účinky hlášené po uvedení na trh: Frekvence není známa: hemolýza, anafylaktický šok, tranzitorní ischemická ataka, cévní mozková příhoda, infarkt myokardu, hypotenze, hluboká žilní trombóza, plicní embolie, plicní edém, pozitivní přímý Coombsův test, snížená saturace kyslíkem, akutní plicní poranění v souvislosti s transfuzí.
Uchovávání: Uchovávejte při teplotě do 25 °C. Chraňte před mrazem. Uchovávejte vnitřní obal v krabičce, aby byl přípravek chráněn před světlem. Držitel rozhodnutí o registraci: Takeda Manufacturing Austria AG, Industriestrasse 67, A-1221 Vídeň, Rakousko. Registrační čísla: EU/1/05/329/001-006. Poslední revize SPC: 05/2020.
*Všimněte si, prosím, změn v informacích o léčivém přípravku
Dříve než začnete přípravek předepisovat, seznamte se, prosím, s úplným souhrnem údajů o přípravku (SPC). Výdej léčivého přípravku je vázán na lékařský předpis. Léčivý přípravek je hrazen z prostředků veřejného zdravotního pojištění. Úplné znění SPC naleznete na www.sukl.cz.
Žádáme zdravotnické pracovníky, aby hlásili podezření na nežádoucí účinky na SÚKL nebo společnosti Takeda emailem na drugsafety-cz@takeda.com.