Hemofilické dni 2022 – přednáška doc. Jana Blatného, FN Brno
„Léčbu hemofilie umíme ušít na míru.“ Přednáška doc. MUDr. Jana Blatného, Ph.D., primáře Oddělení dětské hematologie a biochemie Dětské nemocnice FN Brno, jež byla v programu XIII. hemofilických dní v Bratislavě součástí bloku věnovaného komplexní péči o nemocné s krvácivými chorobami.



ADVATE
Zkrácené informace o léčivém přípravku
ADVATE 250 IU prášek a rozpouštědlo pro injekční roztok, ADVATE 500 IU prášek a rozpouštědlo pro injekční roztok, ADVATE 1000 IU prášek a rozpouštědlo pro injekční roztok, ADVATE 1500 IU prášek a rozpouštědlo pro injekční roztok, ADVATE 2000 IU prášek a rozpouštědlo pro injekční roztok, ADVATE 3000 IU prášek a rozpouštědlo pro injekční roztok
Dříve než začnete přípravek předepisovat, seznamte se, prosím, s úplným souhrnem údajů o přípravku (SPC).
Složení: Léčivá látka: Prášek: 250/500/1000/1500/2000/3000 IU lidského koagulačního faktoru VIII (rDNA), octocogum alfa, Rozpouštědlo 5 ml: Po rekonstituci obsahuje přípravek ADVATE přibližně 50/100/200/300/400/600 IU/ml lidského koagulačního faktoru VIII (rDNA), octocogum alfa.
Prášek 250/500/1000/1500 IU lidského koagulačního faktoru VIII (rDNA), octocogum alfa, Rozpouštědlo 2 ml: Po rekonstituci obsahuje přípravek ADVATE přibližně 125/250/500/750 IU/ml lidského koagulačního faktoru VIII (rDNA), octocogum alfa.
Indikace: Léčba a profylaxe krvácení u pacientů s hemofilií A (vrozený nedostatek faktoru VIII).
Dávkování a způsob podání: Léčba on demand (dle potřeby): Výpočet požadované dávky faktoru VIII je založen na empirickém zjištění, že 1 IU faktoru VIII na 1 kg tělesné hmotnosti zvyšuje aktivitu plazmatického faktoru VIII o 2 IU/dl. Dávka se určuje podle následujícího vzorce: Požadované jednotky (IU) = tělesná hmotnost (kg) x požadovaný vzestup faktoru VIII (%) x 0,5. Profylaxe: Pro dlouhodobou profylaxi krvácení u pacientů s těžkou hemofilií A jsou obvyklé dávky 20 až 40 IU faktoru VIII na 1 kg tělesné hmotnosti v intervalu dvou až tří dnů. U pacientů mladších 6 let se k profylaktické léčbě doporučují dávky faktoru VIII 20 až 50 IU na kg tělesné hmotnosti 3krát až 4krát týdně. Způsob podání: ADVATE má být podáván intravenózně. Kontraindikace: Hypersenzitivita na léčivou látku nebo na kteroukoli pomocnou látku nebo na myší nebo křeččí proteiny.
Upozornění: Hypersenzitivita: U přípravku ADVATE byly hlášeny hypersenzitivní reakce alergického typu (včetně anafylaxe). Přípravek obsahuje stopy myších a křeččích proteinů. Pokud se symptomy hypersenzitivity objeví, pacienti musí ihned přerušit používání přípravku a kontaktovat svého lékaře. Inhibitory: Tvorba neutralizujících protilátek (inhibitorů) faktoru VIII je známou komplikací léčby jedinců s hemofilií A. Riziko vzniku inhibitorů souvisí se závažností onemocnění i s expozicí faktoru VIII, přičemž toto riziko je nejvyšší během prvních 20 dnů expozice. Obecně platí, že všichni pacienti léčení přípravky s koagulačním faktorem VIII musí být pečlivě sledováni s ohledem na vznik inhibitorů pomocí příslušných klinických pozorování a laboratorních testů. Důrazně se doporučuje po každém podání přípravku ADVATE pacientovi zaznamenat název a číslo šarže přípravku, aby bylo možné přesně dohledat, jaká šarže léčivého přípravku byla pacientovi podána.
Významné interakce: Nebyly provedeny žádné studie interakce s přípravkem ADVATE. Studie kompatibility nejsou k dispozici, a proto nesmí být tento léčivý přípravek mísen s jinými léčivými přípravky nebo rozpouštědly. Hlavní nežádoucí účinky: Klinické studie s přípravkem ADVATE zahrnovaly 418 pacientů s alespoň jednou expozicí přípravku ADVATE; bylo při nich nahlášeno celkem 93 nežádoucích účinků léčiva (NÚ). NÚ, které se vyskytovaly s nejvyšší frekvencí, byly vývoj neutralizujících protilátek faktoru VIII (inhibitorů), bolest hlavy a horečka. Alergické reakce nebo hypersenzitivní reakce byly pozorovány vzácně a mohou v některých případech progredovat do těžké anafylaxe (včetně šoku). Může být pozorován vývoj protilátek proti myšímu a/nebo křeččímu proteinu se souvisejícími hypersenzitivními reakcemi. K rozvoji neutralizujících protilátek (inhibitorů) může dojít u pacientů s hemofilií A, kteří jsou léčeni faktorem VIII, včetně přípravku ADVATE. Pokud se takové inhibitory objeví, projeví se tento stav jako nedostatečná klinická odpověď.
Uchovávání: Uchovávejte v chladničce (2°C – 8°C). Chraňte před mrazem. Během doby použitelnosti může být přípravek uchováván při pokojové teplotě (do 25°C) po jedno období nepřesahující 6 měsíců. Přípravek nesmí být vrácen zpět do chladničky.
Držitel rozhodnutí o registraci: Takeda Manufacturing Austria AG, Industriestrasse 67, A-1221 Vídeň, Rakousko.
Registrační čísla: EU/1/03/271/001-020.
Poslední revize SPC: 02/2021.
Výdej léčivého přípravku je vázán na lékařský předpis.
Léčivý přípravek je hrazen z prostředků veřejného zdravotního pojištění.
Úplné znění SPC naleznete na www.sukl.cz.
Žádáme zdravotnické pracovníky, aby hlásili podezření na nežádoucí účinky na SÚKL nebo společnosti Takeda emailem na AE.CZE@takeda.com. Podezření na nežádoucí účinky hlaste také podle národních legislativních požadavků.
Dříve než začnete přípravek předepisovat, seznamte se, prosím, s úplným souhrnem údajů o přípravku (SPC).
Složení: Léčivá látka: Prášek: 250/500/1000/1500/2000/3000 IU lidského koagulačního faktoru VIII (rDNA), octocogum alfa, Rozpouštědlo 5 ml: Po rekonstituci obsahuje přípravek ADVATE přibližně 50/100/200/300/400/600 IU/ml lidského koagulačního faktoru VIII (rDNA), octocogum alfa.
Prášek 250/500/1000/1500 IU lidského koagulačního faktoru VIII (rDNA), octocogum alfa, Rozpouštědlo 2 ml: Po rekonstituci obsahuje přípravek ADVATE přibližně 125/250/500/750 IU/ml lidského koagulačního faktoru VIII (rDNA), octocogum alfa.
Indikace: Léčba a profylaxe krvácení u pacientů s hemofilií A (vrozený nedostatek faktoru VIII).
Dávkování a způsob podání: Léčba on demand (dle potřeby): Výpočet požadované dávky faktoru VIII je založen na empirickém zjištění, že 1 IU faktoru VIII na 1 kg tělesné hmotnosti zvyšuje aktivitu plazmatického faktoru VIII o 2 IU/dl. Dávka se určuje podle následujícího vzorce: Požadované jednotky (IU) = tělesná hmotnost (kg) x požadovaný vzestup faktoru VIII (%) x 0,5. Profylaxe: Pro dlouhodobou profylaxi krvácení u pacientů s těžkou hemofilií A jsou obvyklé dávky 20 až 40 IU faktoru VIII na 1 kg tělesné hmotnosti v intervalu dvou až tří dnů. U pacientů mladších 6 let se k profylaktické léčbě doporučují dávky faktoru VIII 20 až 50 IU na kg tělesné hmotnosti 3krát až 4krát týdně. Způsob podání: ADVATE má být podáván intravenózně. Kontraindikace: Hypersenzitivita na léčivou látku nebo na kteroukoli pomocnou látku nebo na myší nebo křeččí proteiny.
Upozornění: Hypersenzitivita: U přípravku ADVATE byly hlášeny hypersenzitivní reakce alergického typu (včetně anafylaxe). Přípravek obsahuje stopy myších a křeččích proteinů. Pokud se symptomy hypersenzitivity objeví, pacienti musí ihned přerušit používání přípravku a kontaktovat svého lékaře. Inhibitory: Tvorba neutralizujících protilátek (inhibitorů) faktoru VIII je známou komplikací léčby jedinců s hemofilií A. Riziko vzniku inhibitorů souvisí se závažností onemocnění i s expozicí faktoru VIII, přičemž toto riziko je nejvyšší během prvních 20 dnů expozice. Obecně platí, že všichni pacienti léčení přípravky s koagulačním faktorem VIII musí být pečlivě sledováni s ohledem na vznik inhibitorů pomocí příslušných klinických pozorování a laboratorních testů. Důrazně se doporučuje po každém podání přípravku ADVATE pacientovi zaznamenat název a číslo šarže přípravku, aby bylo možné přesně dohledat, jaká šarže léčivého přípravku byla pacientovi podána.
Významné interakce: Nebyly provedeny žádné studie interakce s přípravkem ADVATE. Studie kompatibility nejsou k dispozici, a proto nesmí být tento léčivý přípravek mísen s jinými léčivými přípravky nebo rozpouštědly. Hlavní nežádoucí účinky: Klinické studie s přípravkem ADVATE zahrnovaly 418 pacientů s alespoň jednou expozicí přípravku ADVATE; bylo při nich nahlášeno celkem 93 nežádoucích účinků léčiva (NÚ). NÚ, které se vyskytovaly s nejvyšší frekvencí, byly vývoj neutralizujících protilátek faktoru VIII (inhibitorů), bolest hlavy a horečka. Alergické reakce nebo hypersenzitivní reakce byly pozorovány vzácně a mohou v některých případech progredovat do těžké anafylaxe (včetně šoku). Může být pozorován vývoj protilátek proti myšímu a/nebo křeččímu proteinu se souvisejícími hypersenzitivními reakcemi. K rozvoji neutralizujících protilátek (inhibitorů) může dojít u pacientů s hemofilií A, kteří jsou léčeni faktorem VIII, včetně přípravku ADVATE. Pokud se takové inhibitory objeví, projeví se tento stav jako nedostatečná klinická odpověď.
Uchovávání: Uchovávejte v chladničce (2°C – 8°C). Chraňte před mrazem. Během doby použitelnosti může být přípravek uchováván při pokojové teplotě (do 25°C) po jedno období nepřesahující 6 měsíců. Přípravek nesmí být vrácen zpět do chladničky.
Držitel rozhodnutí o registraci: Takeda Manufacturing Austria AG, Industriestrasse 67, A-1221 Vídeň, Rakousko.
Registrační čísla: EU/1/03/271/001-020.
Poslední revize SPC: 02/2021.
Výdej léčivého přípravku je vázán na lékařský předpis.
Léčivý přípravek je hrazen z prostředků veřejného zdravotního pojištění.
Úplné znění SPC naleznete na www.sukl.cz.
Žádáme zdravotnické pracovníky, aby hlásili podezření na nežádoucí účinky na SÚKL nebo společnosti Takeda emailem na AE.CZE@takeda.com. Podezření na nežádoucí účinky hlaste také podle národních legislativních požadavků.
Vše o hemofilii
Společnost Takeda je tradičním partnerem pacientů s hemofilií i zdravotníků, kteří o ně pečují. V České republice mohou hemofilici i lékaři profitovat z našich:
- dlouhodobých zkušeností,
- intenzivního výzkumu,
- moderních léčivých přípravků a technologií, které přinášíme na trh.
Zaměřujeme se také na optimalizaci léčby, zvyšování komfortu jejího podávání i spolupráci s odbornými společnostmi a pacientskými organizacemi. Jsme připraveni reagovat na výzvy, které léčba hematologických onemocnění přináší. V této oblasti disponujeme rozsáhlým portfoliem léčivých přípravků, zahrnujícím plazmatické, rekombinantní produkty i léčiva s prolongovaným působením.
Přinášíme sofistikovaná léčiva i pomůcky pro léčbu hemofilie všech typů a věříme, že se nám i s Vaší pomocí podaří naplnit naši vizi Bleed-Free World a že se „svět bez krvácení“ v budoucnu skutečně stane realitou. Vyzkoušejte se svými pacienty první spojení webové a mobilní aplikace v léčbě hemofilie a jediný schválený zdravotnický prostředek k individuálnímu nastavení optimální profylaxe hemofilika – webovou a mobilní aplikaci myPKFiT.
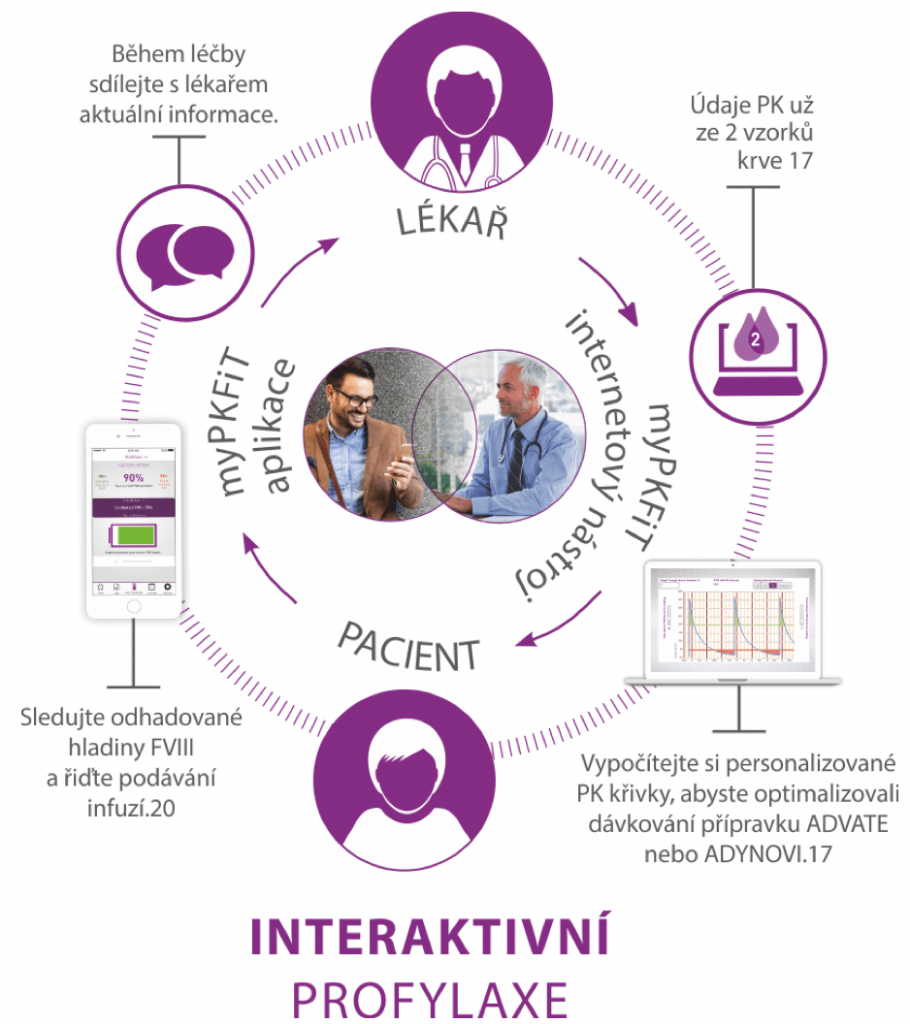
Používání aplikace myPKFiT umožňuje sdílet všechny aktualizace s ošetřujícím lékařem, optimalizovat dávkování na základě osobního farmakokinetického profilu a zároveň zvyšovat osobní účast na léčbě a samostatnost v jejím managementu. I v ČR se toto řešení setkalo s velmi příznivým ohlasem u odborníků i pacientů a velké množství z nich již výhod této technologie naplno využívá. Buďte dalšími z nich.
Stručně o hemofilii
Hemofilie je vzácná, obvykle dědičná koagulopatie. Jedná se o gonosomálně recesivní onemocnění. Defektní gen je lokalizován na pohlavním chromosomu X, proto se hemofilie vyskytuje prakticky jen u mužů, u žen bývá extrémně vzácná. Hemofilie se projevuje poruchou srážlivosti krve a z toho plynoucím krvácením. Zdaleka nejčastějším typem je hemofilie typu A (deficit faktoru VIII). Druhým nejčastějším typem je typ B (deficit faktoru IX). Existují i jiná krvácivá onemocnění s deficity jiných srážecích faktorů, která se obvykle již jako hemofilie neoznačují.
Hemofilii lze odhalit již u batolat, kdy dítě začíná chodit a také padat. Ke krvácení dochází i při minimálním nárazu nebo zcela bez příčiny. Na exponovaných místech (hlava, hýždě, končetiny) se pak objevují typické podkožní a někdy i svalové hematomy. Po pádu na obličej nezřídka dochází
i ke krvácení v dutině ústní. Tyto projevy může okolí rodiny mylně vyhodnotit jako týrání dítěte.
V novorozeneckém věku je krvácení poměrně vzácné. U mladších dětí a školáků může frekvenci krvácení zesílit i stres, například v období zkoušek. Pozoruje se i sezónní vliv na krvácení, například na jaře a na podzim.
Hemofilii rozdělujeme v závislosti na množství chybějícího koagulačního faktoru na:
– Těžkou formu (< 1 IU/dl nebo < 1 % normální koncentrace srážecího faktoru), při které dochází k častým spontánním krvácením především do velkých kloubů nebo svalů.1
– Středně těžkou formu (1–5 % normální koncentrace srážecího faktoru). Krvácení vzniká již po nepatrných úrazech nebo poraněních, může vzniknout i spontánně.
– Lehkou formu (> 5–40 % normální koncentrace srážecího faktoru). Ke dlouhotrvajícímu krvácení dochází po stomatologických a operačních výkonech či úrazech, ale pacient nekrvácí spontánně do kloubů nebo dalších částí těla.1
C-APROM/CZ//0701
Literatura
- Srivastava A, Brewer AK, Mauser-Bunschoten EP, et al.; Treatment Guidelines Working Group on Behalf of The World Federation Of Hemophilia. Guidelines for the management of hemophilia. Haemophilia 2013; 19 (1): e1–e47.
- Blanchette VS, Key NS, Ljung LR, et al.; Subcommittee on Factor VIII, Factor IX and Rare Coagulation Disorders of the Scientific and Standardization Committee of the International Society on Thrombosis and Hemostasis. Definitions in hemophilia: communication from the SSC of the ISTH. J Thromb Haemost 2014; 12 (11): 1935–1939.
- Rodriguez-Merchan EC. Musculoskeletal complications of hemophilia. HSS J 2010; 6 (1): 37–42.
- Aledort LM, Haschmeyer RH, Pettersson H. A longitudinal study of orthopaedic outcomes for severe factor-VIII deficient haemophiliacs. The Orthopaedic Outcome Study Group. J Intern Med 1994; 236 (4): 391–399.
- National Hemophilia Foundation. MASAC recommendation concerning prophylaxis (regular administration of clotting factor concentrate to prevent bleeding). MASAC Document #241. February 28, 2016.
- Oldenburg J, Zimmermann R, Katsarou O, et al. Controlled, cross-sectional MRI evaluation of joint status in severe haemophilia A patients treated with prophylaxis vs. on demand. Haemophilia 2015; 21 (2): 171–179.
- Pasca S, Milan M, Sarolo L, Zanon E. PK-driven prophylaxis versus standard prophylaxis: When a tailored treatment may be a real and achievable cost-saving approach in children with severe hemophilia A. Thromb Res 2017; 157: 58–63.
- Údaje z prospektivní klinické studie fáze IV s Advate (n = 66).
- Iorio A, Marcucci M, Cheng J, et al. Patient data meta-analysis of Post-Authorization Safety Surveillance (PASS) studies of haemophilia A patients treated with rAHF-PFM. Haemophilia 2014; 20 (6): 777–783.
- Pět poregistračních studií (PASS) s Advate.
- Mahdi AJ, Obaji SG, Collins PW. Role of enhanced halife factor VIII and IX in the treatment of haemophilia. Br J Haematol 2015; 169 (6): 768–776.
- Konkle BA, Stasyshyn O, Chowdary P, et al. Pegylated, full-length, recombinant factor VIII for prophylactic and on-demand treatment of severe hemophilia A. Blood 2015; 126 (9): 1078–1085.
- Valentino LA. Considerations in individualizing prophylaxis in patients with haemophilia A. Haemophilia 2014; 20 (5): 607–615.
- Windyga J, Zbikowski P, Ambroziak P, et al. Management of factor VII-deficient patients undergoing joint surgeries–preliminary results of locally developed treatment regimen. Haemophilia 2013; 19 (1): 89–93.
- Urasinski T, Stasyshyn O, Andreeva T, et al. Recombinant factor IX (BAX326) in previously treated paediatric patients with haemophilia B: a prospective clinical trial. Haemophilia 2015; 21 (2): 196–203.
- Gouw SC, van der Bom JG, van den Berget HM. Treatment-related risk factors of inhibitor development in previously untreated patients with hemophilia A: the CANAL cohort study. Blood 2007; 109 (11): 4648–4654.
- Coppola A, Di Capua M, Di Minno MN, et al. Treatment of hemophilia: a review of current advances and ongoing issues. J Blood Med 2010; 1: 183–195.
- Morfini M, Auerswald G, Kobelt RA, et al. Prophylactic treatment of haemophilia patients with inhibitors: clinical experience with recombinant factor VIIa in European Haemophilia Centres. Haemophilia 2007; 13 (5): 502–507.
- Doc. MUDr. Zuzana Čermáková, Ph.D. – Centrum pro trombózu a hemostázu, hemofilické centrum, Krevní centrum FN Ostrava, Farmakologický ústav, LF MU v Brně.
- Čermáková Z, Hrdličková R, Smejkal P, et al.; Pracovní skupina ČNHP pro standardy. Diagnostika a léčba získané hemofilie – konsenzuální doporučení Českého národního hemofilického programu (ČNHP). Transfuze Hematol dnes 2017; 23 (2): 101–109.
Nové výzvy v léčbě chronické hypoparatyreózy
Společnost Takeda dne 25.3.2021 připravila on-line edukační seminář NOVÉ VÝZVY V LÉČBĚ CHRONICKÉ HYPOPARATYREÓZY, jehož cílem bylo sdílení zkušeností týkající se diagnostiky a léčby chronické hypoparatyreózy.
Pozvání na seminář přijala Dr. Karin Amrein (Rakousko), která má bohaté klinické zkušenosti s léčbou rhPTH (1-84) a prof. Michal Kršek, který je jedním z autorů Doporučených postupů pro diagnostiku a léčbu chronické hypoparatyreózy.
Více informací pro Vaše pacienty www.hypopara.cz.
![]()
Mohlo by Vás zajímat




NATPAR
Zkrácené informace o léčivém přípravku
▼ Tento léčivý přípravek podléhá dalšímu sledování. To umožní rychlé získání nových informací o bezpečnosti. Žádáme zdravotnické pracovníky, aby hlásili jakákoli podezření na nežádoucí účinky. Podrobnosti o hlášení nežádoucích účinků viz bod 4.8 SPC.
Název přípravku: Natpar 25 mikrogramů/dávka prášek a rozpouštědlo pro injekční roztok; Natpar 50 mikrogramů/dávka prášek a rozpouštědlo pro injekční roztok; Natpar 75 mikrogramů/dávka prášek a rozpouštědlo pro injekční roztok; Natpar 100 mikrogramů/dávka prášek a rozpouštědlo pro injekční Složení: Jedna dávka obsahuje po rekonstituci 25/50/75/100 mikrogramů hormonum parathyroidum (rDNA)* v 71,4 mikrolitrech roztoku. Jedna zásobní vložka obsahuje 350/700/1050/1400 mikrogramů hormonum parathyroidum (rDNA). *Parathormon (rDNA) produkovaný E. coli pomocí rekombinantní DNA technologie je identický se sekvencí 84 aminokyselin endogenního lidského parathormonu. Indikace: Přípravek Natpar je indikován jako přídatná léčba dospělých pacientů s chronickou hypoparathyreózou, kteří nejsou dostatečně kontrolováni samostatnou standardní léčbou. Dávkování a způsob podání: Zahajte léčbu 50 mikrogramy jednou denně subkutánní injekcí do stehna (střídejte stehna každý den). Pokud je před podáním dávky sérové kalcium > 2,25 mmol/l, je možné zvážit podání počáteční dávky 25 mikrogramů. U pacientů užívajících aktivní vitamin D snižte jeho dávku o 50 %, pokud je sérové kalcium před podáním dávky nad 1,87 mmol/l. Udržujte doplňkovou dávku kalcia. Změřte sérovou koncentraci kalcia během 2 až 5 dnů před podáním dávky. Pokud je sérová hladina kalcia před dávkou pod 1,87 mmol/l nebo nad 2,55 mmol/l, mělo by se měření opakovat následující den. Upravte dávku aktivního vitamínu D nebo doplňku kalcia nebo obou na základě sérové hladiny kalcia a klinického hodnocení. Úpravy se mají opakovat, dokud nebudou cílové hladiny sérového kalcia před dávkou v rozsahu 2,0 – 2,25 mmol/l, aktivní vitamín D bude ukončen a doplněk kalcia je dostatečný pro splnění denních požadavků. Během titrace musí být monitorována koncentrace sérového kalcia. Dávka přípravku Natpar se může zvýšit o 25 mikrogramů přibližně každé 2 až 4 týdny, do maximální denní dávky 100 mikrogramů. K titraci dávky dolů na minimum 25 mikrogramů může dojít kdykoliv. Pokyny k úpravě dávky po úvodním období – viz SPC. Přípravek Natpar je vhodný pro podávání samotným pacientem jako subkutánní injekce jednou denně střídavě do každého stehna. Kontraindikace: Hypersenzitivita na léčivou látku nebo na kteroukoli pomocnou látku. U pacientů, kteří podstupují nebo dříve podstupovali radioterapii skeletu, s malignitami skeletu nebo kostními metastázami, kteří mají zvýšené výchozí riziko vzniku osteosarkomu (pacienti s Pagetovou chorobou kostí nebo dědičnými poruchami), s nevysvětleným zvýšením kostní alkalické fosfatázy a s pseudohypoparathyreózou. Zvláštní upozornění a opatření pro použití: Monitorování pacientů během léčby: Sérové hladiny kalcia před dávkou a v některých případech po dávce musí být během léčby přípravkem Natpar sledovány. Hyperkalcemie: často se objevovala během titračního období. V případě závažné hyperkalcemie (> 3,0 mmol/l nebo nad horní hranici normy s příznaky), zvažte hydrataci a dočasné zastavení podávání přípravku Natpar, kalcia a aktivního vitamínu D, dokud se sérové kalcium nevrátí do normálního rozsahu. Hypokalcemie: Riziko závažné hypokalcemie je nejvyšší po odložení podání, vynechání nebo náhlém vysazení přípravku Natpar, může se ale vyskytnout kdykoli. Souběžné použití se srdečními glykosidy: Hyperkalcemie z jakékoliv příčiny může predisponovat k toxicitě digitalis. Závažná porucha funkce ledvin nebo jater: Používat s opatrností u pacientů se závažnou poruchou ledvin nebo jater. Použití u mladých dospělých: Používat s opatrností u mladých dospělých pacientů s otevřenými epifýzami, protože tito pacienti mohou být vystaveni zvýšenému riziku vzniku osteosarkomu. Použití u starších pacientů: Klinické studie nezahrnovaly dostatečný počet subjektů ve věku ≥ 65 let pro stanovení, zda je odpověď u těchto subjektů odlišná od mladších subjektů. Tachyfylaxe: Účinek přípravku Natpar se může po čase u některých pacientů snížit. Urolitiáza: U pacientů s aktivní nebo nedávnou urolitiázou je nutno používat přípravek Natpar s opatrností vzhledem k jeho potenciálu tento stav zhoršovat. Hypersenzitivita: Po uvedení na trh byly hlášeny hypersenzitivní reakce, které mohou zahrnovat anafylaxi, dyspnoi, angioedém, kopřivku, vyrážku atd. Interakce: Kombinované použití přípravku Natpar a srdečních glykosidů může pacienty predisponovat k toxicitě digitalisu, pokud vznikne hyperkalcémie. Současné používání přípravku Natpar s bisfosfonáty se nedoporučuje. Fertilita, těhotenství a kojení: Údaje o podávání přípravku Natpar těhotným ženám nejsou k dispozici. Studie reprodukční toxicity na zvířatech nenaznačují přímé nebo nepřímé škodlivé účinky. Není známo, zda se přípravek Natpar vylučuje do lidského mateřského mléka. Dostupné farmakologické údaje u zvířat prokázaly vylučování přípravku Natpar do mléka. Neexistují žádné údaje u člověka o vlivu přípravku Natpar na plodnost. Studie na zvířatech nenaznačují žádnou poruchu plodnosti. Nežádoucí účinky: Nejčastější nežádoucí reakce byly hyperkalcemie, hypokalcemie a jejich související klinické projevy, zahrnující bolesti hlavy, průjem, zvracení, parestezie, hypestezie a hyperkalciurie. Ostatní nežádoucí účinky – viz SPC. Předávkování: Může způsobit hyperkalcémii. Těžká hyperkalcémie může být život ohrožující stav vyžadující neodkladnou lékařskou péči a pečlivé monitorování. Podmínky uchovávání: Uchovávejte v chladničce (2 °C – 8 °C). Chraňte před mrazem. Uchovávejte zásobní vložky v držáku v krabičce, aby byly chráněny před světlem. Podmínky uchovávání po rekonstituci – viz SPC. Držitel rozhodnutí o registraci: Shire Pharmaceuticals Ireland Limited, Block 2 & 3 Miesian Plaza, 50 – 58 Baggot Street Lower, Dublin 2, Irsko. Registrační čísla: EU/1/15/1078/001-004. Poslední revize SPC: 04/2020.
Výdej léčivého přípravku je vázán na lékařský předpis. Léčivý přípravek není hrazen z prostředků veřejného zdravotního pojištění. Před předepsáním se seznamte s úplným zněním Souhrnu údajů o přípravku.
Žádáme zdravotnické pracovníky, aby hlásili podezření na nežádoucí účinky na SÚKL nebo společnosti Takeda emailem na AE.CZE@takeda.com. Podezření na nežádoucí účinky hlaste také podle národních legislativních požadavků.
▼ Tento léčivý přípravek podléhá dalšímu sledování. To umožní rychlé získání nových informací o bezpečnosti. Žádáme zdravotnické pracovníky, aby hlásili jakákoli podezření na nežádoucí účinky. Podrobnosti o hlášení nežádoucích účinků viz bod 4.8 SPC.
Název přípravku: Natpar 25 mikrogramů/dávka prášek a rozpouštědlo pro injekční roztok; Natpar 50 mikrogramů/dávka prášek a rozpouštědlo pro injekční roztok; Natpar 75 mikrogramů/dávka prášek a rozpouštědlo pro injekční roztok; Natpar 100 mikrogramů/dávka prášek a rozpouštědlo pro injekční Složení: Jedna dávka obsahuje po rekonstituci 25/50/75/100 mikrogramů hormonum parathyroidum (rDNA)* v 71,4 mikrolitrech roztoku. Jedna zásobní vložka obsahuje 350/700/1050/1400 mikrogramů hormonum parathyroidum (rDNA). *Parathormon (rDNA) produkovaný E. coli pomocí rekombinantní DNA technologie je identický se sekvencí 84 aminokyselin endogenního lidského parathormonu. Indikace: Přípravek Natpar je indikován jako přídatná léčba dospělých pacientů s chronickou hypoparathyreózou, kteří nejsou dostatečně kontrolováni samostatnou standardní léčbou. Dávkování a způsob podání: Zahajte léčbu 50 mikrogramy jednou denně subkutánní injekcí do stehna (střídejte stehna každý den). Pokud je před podáním dávky sérové kalcium > 2,25 mmol/l, je možné zvážit podání počáteční dávky 25 mikrogramů. U pacientů užívajících aktivní vitamin D snižte jeho dávku o 50 %, pokud je sérové kalcium před podáním dávky nad 1,87 mmol/l. Udržujte doplňkovou dávku kalcia. Změřte sérovou koncentraci kalcia během 2 až 5 dnů před podáním dávky. Pokud je sérová hladina kalcia před dávkou pod 1,87 mmol/l nebo nad 2,55 mmol/l, mělo by se měření opakovat následující den. Upravte dávku aktivního vitamínu D nebo doplňku kalcia nebo obou na základě sérové hladiny kalcia a klinického hodnocení. Úpravy se mají opakovat, dokud nebudou cílové hladiny sérového kalcia před dávkou v rozsahu 2,0 – 2,25 mmol/l, aktivní vitamín D bude ukončen a doplněk kalcia je dostatečný pro splnění denních požadavků. Během titrace musí být monitorována koncentrace sérového kalcia. Dávka přípravku Natpar se může zvýšit o 25 mikrogramů přibližně každé 2 až 4 týdny, do maximální denní dávky 100 mikrogramů. K titraci dávky dolů na minimum 25 mikrogramů může dojít kdykoliv. Pokyny k úpravě dávky po úvodním období – viz SPC. Přípravek Natpar je vhodný pro podávání samotným pacientem jako subkutánní injekce jednou denně střídavě do každého stehna. Kontraindikace: Hypersenzitivita na léčivou látku nebo na kteroukoli pomocnou látku. U pacientů, kteří podstupují nebo dříve podstupovali radioterapii skeletu, s malignitami skeletu nebo kostními metastázami, kteří mají zvýšené výchozí riziko vzniku osteosarkomu (pacienti s Pagetovou chorobou kostí nebo dědičnými poruchami), s nevysvětleným zvýšením kostní alkalické fosfatázy a s pseudohypoparathyreózou. Zvláštní upozornění a opatření pro použití: Monitorování pacientů během léčby: Sérové hladiny kalcia před dávkou a v některých případech po dávce musí být během léčby přípravkem Natpar sledovány. Hyperkalcemie: často se objevovala během titračního období. V případě závažné hyperkalcemie (> 3,0 mmol/l nebo nad horní hranici normy s příznaky), zvažte hydrataci a dočasné zastavení podávání přípravku Natpar, kalcia a aktivního vitamínu D, dokud se sérové kalcium nevrátí do normálního rozsahu. Hypokalcemie: Riziko závažné hypokalcemie je nejvyšší po odložení podání, vynechání nebo náhlém vysazení přípravku Natpar, může se ale vyskytnout kdykoli. Souběžné použití se srdečními glykosidy: Hyperkalcemie z jakékoliv příčiny může predisponovat k toxicitě digitalis. Závažná porucha funkce ledvin nebo jater: Používat s opatrností u pacientů se závažnou poruchou ledvin nebo jater. Použití u mladých dospělých: Používat s opatrností u mladých dospělých pacientů s otevřenými epifýzami, protože tito pacienti mohou být vystaveni zvýšenému riziku vzniku osteosarkomu. Použití u starších pacientů: Klinické studie nezahrnovaly dostatečný počet subjektů ve věku ≥ 65 let pro stanovení, zda je odpověď u těchto subjektů odlišná od mladších subjektů. Tachyfylaxe: Účinek přípravku Natpar se může po čase u některých pacientů snížit. Urolitiáza: U pacientů s aktivní nebo nedávnou urolitiázou je nutno používat přípravek Natpar s opatrností vzhledem k jeho potenciálu tento stav zhoršovat. Hypersenzitivita: Po uvedení na trh byly hlášeny hypersenzitivní reakce, které mohou zahrnovat anafylaxi, dyspnoi, angioedém, kopřivku, vyrážku atd. Interakce: Kombinované použití přípravku Natpar a srdečních glykosidů může pacienty predisponovat k toxicitě digitalisu, pokud vznikne hyperkalcémie. Současné používání přípravku Natpar s bisfosfonáty se nedoporučuje. Fertilita, těhotenství a kojení: Údaje o podávání přípravku Natpar těhotným ženám nejsou k dispozici. Studie reprodukční toxicity na zvířatech nenaznačují přímé nebo nepřímé škodlivé účinky. Není známo, zda se přípravek Natpar vylučuje do lidského mateřského mléka. Dostupné farmakologické údaje u zvířat prokázaly vylučování přípravku Natpar do mléka. Neexistují žádné údaje u člověka o vlivu přípravku Natpar na plodnost. Studie na zvířatech nenaznačují žádnou poruchu plodnosti. Nežádoucí účinky: Nejčastější nežádoucí reakce byly hyperkalcemie, hypokalcemie a jejich související klinické projevy, zahrnující bolesti hlavy, průjem, zvracení, parestezie, hypestezie a hyperkalciurie. Ostatní nežádoucí účinky – viz SPC. Předávkování: Může způsobit hyperkalcémii. Těžká hyperkalcémie může být život ohrožující stav vyžadující neodkladnou lékařskou péči a pečlivé monitorování. Podmínky uchovávání: Uchovávejte v chladničce (2 °C – 8 °C). Chraňte před mrazem. Uchovávejte zásobní vložky v držáku v krabičce, aby byly chráněny před světlem. Podmínky uchovávání po rekonstituci – viz SPC. Držitel rozhodnutí o registraci: Shire Pharmaceuticals Ireland Limited, Block 2 & 3 Miesian Plaza, 50 – 58 Baggot Street Lower, Dublin 2, Irsko. Registrační čísla: EU/1/15/1078/001-004. Poslední revize SPC: 04/2020.
Výdej léčivého přípravku je vázán na lékařský předpis. Léčivý přípravek není hrazen z prostředků veřejného zdravotního pojištění. Před předepsáním se seznamte s úplným zněním Souhrnu údajů o přípravku.
Žádáme zdravotnické pracovníky, aby hlásili podezření na nežádoucí účinky na SÚKL nebo společnosti Takeda emailem na AE.CZE@takeda.com. Podezření na nežádoucí účinky hlaste také podle národních legislativních požadavků.