
Oblast léčby hereditárního angioedému
Hereditární angioedém (HAE) se řadí k primárním imunodeficitům, s autosomálně dominantní dědičností. Onemocnění způsobuje nedostatek funkčního C1 inhibitoru (C1-INH) v kontaktním systému a v 75 % se vyskytuje v rodinné anamnéze. Jeho ataky se obvykle vyznačují nepředvídatelnými nebo prodromálně avizovanými otoky končetin, genitálií, trupu, gastrointestinálního traktu, obličeje a hrtanu. Symptomy mohou přetrvávat až 5 dnů a je třeba jej diferenciální diagnostikou dobře odlišit od alergické reakce. Kvalita pacientova života je výrazně snížena a laryngeální příznaky jsou spojeny s významným zvýšením rizika úmrtí.1 Přitom efektivní léčba existuje.
Portfolio společnosti Takeda obsahuje přípravky pro akutní i profylaktickou léčbu tohoto bolestivého, nepříjemného a nebezpečného onemocnění:
![]()
Takhzyro obsahuje lanadelumab – vůbec první lidskou monoklonální protilátku (mAb) k léčbě HAE, umožňující cílenou a dlouhodobou prevenci atak tohoto onemocnění pomocí jediné pacientem podané podkožní injekce 1× za 2 týdny (doporučená léčba je 300 mg každé 2 týdny; u pacientů, kteří jsou při léčbě stabilně bez atak, lze zvažovat snížení dávky na 300 mg každé 4 týdny). Mechanismem účinku je cílená inhibice plazmatického kalikreninu (klíčového regulátoru atak HAE).
 Video s mechanizmem účniku naleznete (můžete zhlédnout) zde
Video s mechanizmem účniku naleznete (můžete zhlédnout) zde
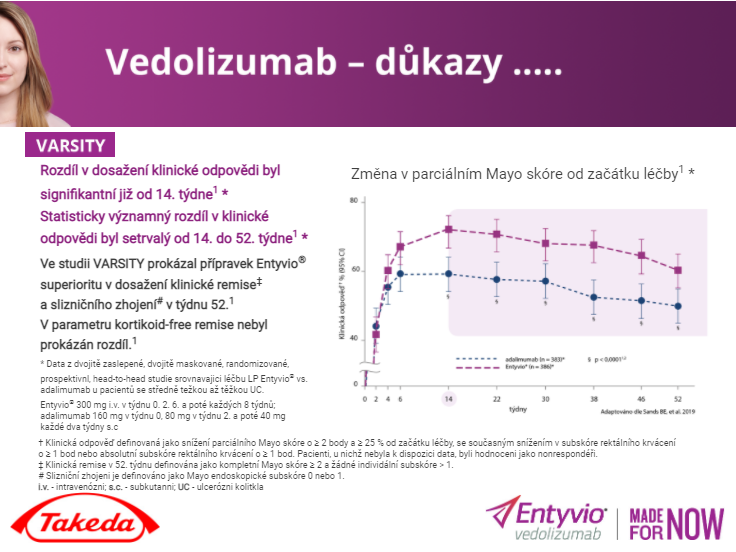
Lék není připravován z lidské plazmy, poločas jeho eliminace je 14 dní a výsledky ukazují, že 87 % pacientů ve studii HELP (n = 125) zaznamenávalo po 6 měsících relativní snížení atak v porovnání s placebovou větví. Dále docházelo i ke snížení závažnosti atak – v sekundárním cíli studie bylo zaznamenáno 83% relativní snížení středních nebo závažných atak v porovnání s placebovou větví. A v post hoc analýze citlivosti mělo 8 z 10 pacientů nulový počet atak po dobu nejméně 4 měsíců. Studie u pacientů zaznamenala také významné snížení strachu z atak, fyzických omezení a únavy.1
 Zhlédněte video prof. Andrea Zanichelli: Shrnutí studie HELP
Zhlédněte video prof. Andrea Zanichelli: Shrnutí studie HELP

Firazyr je prvním a jediným schváleným podkožně podávaným léčivem k akutnímu zaléčení atak HAE samotným pacientem (léčba on demand). Lék se nemusí skladovat v chladničce a je dodáván v předplněné stříkačce. Selektivně inhibuje účinek bradykininu, který zvyšuje vaskulární permeabilitu a vede k tvorbě otoku – zabraňuje tak progresi edému. Během 10 klinických studií bylo tímto lékem léčeno více než 750 nemocných, což odrážejí data v registru a průběžné analýzy (Icatibant Outcome Survey). Nejlepší výsledky léčby byli prokázány při bezprostředním podání do 1 hodiny.1
Pacient by měl mít vždy tuto záchranou léčbu sebou.

Cinryze je z plazmy produkovaný C1 inhibitor indikovaný ke krátkodobé i dlouhodobé profylaxi a on demand léčbě HAE u dospělých a dětí. V České republice není k dispozici.
C-APROM/CZ//0711 Srpen 2020




Vše přehledně na jednom místě

Onkologie

Gastroenterologie
Gastroenterologie

Hematologie

Vzácná onemocnění
Aktuality
Červen 2022




Květen 2022





Nepřehlédněte další zajímavé projekty:



Firazyr 30 mg injekční roztok v předplněné injekční stříkačce
Zkrácené informace o přípravku
Dříve než začnete přípravek předepisovat, seznamte se, prosím, s úplným souhrnem údajů o přípravku (SPC).
Složení: Jedna předplněná injekční stříkačka s objemem 3 ml obsahuje icatibanti acetas odpovídající icatibantum 30 mg. Jeden ml roztoku obsahuje icatibantum 10 mg. Pomocné látky: Chlorid sodný, ledová kyselina octová (k úpravě pH), hydroxid sodný (k úpravě pH), voda pro injekci.
Indikace: Firazyr je indikován k symptomatické léčbě akutních atak dědičného angioedému (HAE) u dospělých, dospívajících a dětí ve věku 2 roky a více, s deficitem inhibitoru esterázy C1.
Dávkování a způsob podání: Firazyr je určen k použití pod vedením zdravotnického pracovníka. Doporučená dávka pro dospělé je jedna subkutánní injekce přípravku Firazyr 30 mg. Ve většině případů stačí k léčbě ataky jediná injekce přípravku Firazyr. Pokud nedojde k úplnému ústupu příznaků nebo se příznaky objeví znovu, po 6 hodinách lze podat druhou injekci přípravku Firazyr. V případě, že ani po podání druhé injekce nedojde k úplnému ústupu příznaků nebo se příznaky objeví znovu, lze po dalších 6 hodinách podat třetí injekci přípravku Firazyr. V průběhu 24 hodin by neměly být podány více než 3 injekce přípravku Firazyr.V rámci klinických studií nebylo podáváno více než 8 injekcí přípravku Firazyr měsíčně. Pediatrická populace: Doporučená dávka přípravku Firazyr na základě tělesné hmotnosti u dětí a dospívajících (ve věku 2 až 17 let) je pro tělesnou hmotnost 12 kg až 25 kg 10 mg (1,0 ml), 26 kg až 40 kg 15 mg (1,5 ml), 41 kg až 50 kg, 20 mg (2,0 ml), 51 kg až 65 kg 25 mg (2,5 ml), > 65 kg 30 mg (3,0 ml). V klinické studii nebyla podána více než 1 injekce přípravku Firazyr na jednu ataku HAE. Starší osoby: Zkušenosti s podáváním přípravku u pacientů ve věku nad 65 let jsou omezené. U starších osob byla prokázána zvýšená systémová expozice ikatibantu. Není známo, zda je tato skutečnost významná ve vztahu k bezpečnosti přípravku Firazyr. Způsob podání: Firazyr je určen k subkutánnímu podání, nejlépe do břišní oblasti. Injekční roztok přípravku Firazyr by měl být injikován pomalu v důsledku objemu, který se podává. Každá stříkačka přípravku Firazyr je určena pouze pro jednorázové použití. O zahájení podávání přípravku Firazyr ošetřující osobou nebo samotným pacientem by měl rozhodnout pouze lékař se zkušenostmi v diagnostice a léčbě dědičného angioedému. Dospělí: Firazyr může být podáván samotným pacientem nebo ošetřující osobou pouze po proškolení v technice subkutánní injekce provedeném zdravotnickým pracovníkem. Děti a dospívající ve věku 2 – 17 let: Firazyr by měla podávat ošetřující osoba pouze po proškolení v technice subkutánní injekce provedeném zdravotnickým pracovníkem.
Kontraindikace: Hypersenzitivita na léčivou látku nebo na kteroukoli pomocnou látku tohoto přípravku.
Zvláštní upozornění: Laryngeální ataky: Pacienty s laryngeálními atakami je třeba po podání injekce pečlivě sledovat ve vhodném zdravotnickém zařízení, dokud lékař nerozhodne, že pacienta lze bez rizika propustit. Ischemická choroba srdeční: V případě ischémie může antagonismus bradykininových receptorů II. typu teoreticky způsobit zhoršení srdeční funkce a snížení průtoku krve koronárními cévami. Při podávání přípravku Firazyr pacientům s akutní ischemickou chorobou srdeční nebo nestabilní anginou pectoris je proto zapotřebí opatrnosti. Mozková příhoda: Ačkoli existují důkazy, které podporují pozitivní vliv blokády B2 receptorů bezprostředně po vzniku mozkové příhody, teoreticky je možné, že by ikatibant mohl oslabit pozitivní neuroprotektivní účinek bradykininu v pozdní fázi. Proto je zapotřebí opatrnosti při podávání ikatibantu pacientům během několika týdnů po vzniku mozkové příhody. Samostatné podávání pacientem nebo ošetřující osobou: Pacientům, kteří Firazyr nikdy dříve nedostali, by měla být první dávka podána ve zdravotnickém zařízení nebo pod dohledem lékaře. Pokud po samostatném podání nebo podání ošetřující osobou nedojde k úplnému ústupu příznaků nebo se příznaky objeví znovu, doporučuje se, aby pacient nebo ošetřující osoba vyhledali lékařskou pomoc. Pro dospělé by se měly následné dávky, které jsou nutné pro stejnou ataku, podat ve zdravotnickém zařízení. Pacienti s laryngeální atakou mají vždy vyhledat lékařskou pomoc a být sledováni ve zdravotnickém zařízení, a to i poté, co si aplikovali injekci v domácím prostředí. Existují omezené zkušenosti s léčbou více než jedné ataky HAE s použitím přípravku Firazyr u pediatrické populace.
Interakce: Neočekávají se žádné farmakokinetické lékové interakce s postižením CYP450. Současné podávání přípravku Firazyr s inhibitory angiotenzin konvertujícího enzymu (ACE) nebylo zkoumáno. ACE inhibitory jsou kontraindikovány u pacientů s dědičným angioedémem vzhledem k možnému zvýšení hladiny bradykininu.
Účinky na schopnost řídit a obsluhovat stroje: Pacientům je třeba doporučit, aby neřídili a neobsluhovali stroje, jestliže se cítí unaveni nebo mají-li závratě v důsledku ataky dědičného angioedému nebo po užití přípravku Firazyr.
Nežádoucí účinky: V klinických studiích použitých pro registraci bylo celkem 999 atak dědičného angioedému léčeno 30 mg Firazyru podanými subkutánně zdravotnickým pracovníkem. Téměř u všech jedinců, kteří byli v rámci klinických studií léčeni podkožně podávaným ikatibantem, se vyskytly reakce v místě podání injekce (charakterizované podrážděním kůže, otokem, bolestí, svěděním, erytémem, pocitem pálení). Tyto reakce byly zpravidla mírné až středně závažné, přechodné a vymizely bez nutnosti další intervence. Velmi časté (≥1/10): Reakce v místě injekce. Časté (≥1/100 až <1/10): Závrať, bolest hlavy, nauzea, vyrážka, erytém, pruritus, pyrexie, zvýšení transamináz. Není známo: Kopřivka. Pediatrická populace: Celkem 32 pediatrických pacientů s HAE bylo během klinických studií vystaveno léčbě ikatibantem. Přípravek Firazyr byl podán podkožní injekcí v dávce 0,4 mg/kg na základě tělesné hmotnosti do maximální dávky 30 mg.Většina pacientů, kteří byli léčeni podkožním ikatibantem měli reakci v místě injekce, jako je erytém, otok, pocit pálení, bolesti kůže a svědění nebo pruritus. Tyto reakce byly mírné až střední závažnosti a odpovídaly reakcím, které byly hlášené u dospělých. Dva pediatričtí pacienti měli reakci v místě injekce, která byla hodnocena jako závažná a zcela ustoupila během 6 hodin. Tyto reakce zahrnovaly erytém, otok, pálení a pocit tepla. Další informace o nežádoucích účincích jsou uvedeny v úplném znění Souhrnu údajů o přípravku.
Uchovávání: Uchovávejte při teplotě do 25 ○C. Chraňte před mrazem.
Obsah balení: Jedna předplněná injekční stříkačka s jednou jehlou nebo vícečetné balení obsahující tři předplněné injekční stříkačky se třemi jehlami. Na trhu nemusí být všechny velikosti balení.
Držitel rozhodnutí o registraci: Takeda Pharmaceuticals International AG Ireland Branch, Block 3 Miesian Plaza, 50 – 58 Baggot Street Lower, Dublin 2, D02 Y754, Irsko
Registrační čísla: EU/1/08/461/001, EU/1/08/461/002
Poslední revize SPC: 10/2021
Výdej léčivého přípravku je vázán na lékařský předpis.
Léčivý přípravek je hrazen z prostředků veřejného zdravotního pojištění.
Úplné znění SPC naleznete na www.sukl.cz.
Žádáme zdravotnické pracovníky, aby hlásili podezření na nežádoucí účinky na SÚKL nebo společnosti Takeda emailem na AE.CZE@takeda.com. Podezření na nežádoucí účinky hlaste také podle národních legislativních požadavků.
Dříve než začnete přípravek předepisovat, seznamte se, prosím, s úplným souhrnem údajů o přípravku (SPC).
Složení: Jedna předplněná injekční stříkačka s objemem 3 ml obsahuje icatibanti acetas odpovídající icatibantum 30 mg. Jeden ml roztoku obsahuje icatibantum 10 mg. Pomocné látky: Chlorid sodný, ledová kyselina octová (k úpravě pH), hydroxid sodný (k úpravě pH), voda pro injekci.
Indikace: Firazyr je indikován k symptomatické léčbě akutních atak dědičného angioedému (HAE) u dospělých, dospívajících a dětí ve věku 2 roky a více, s deficitem inhibitoru esterázy C1.
Dávkování a způsob podání: Firazyr je určen k použití pod vedením zdravotnického pracovníka. Doporučená dávka pro dospělé je jedna subkutánní injekce přípravku Firazyr 30 mg. Ve většině případů stačí k léčbě ataky jediná injekce přípravku Firazyr. Pokud nedojde k úplnému ústupu příznaků nebo se příznaky objeví znovu, po 6 hodinách lze podat druhou injekci přípravku Firazyr. V případě, že ani po podání druhé injekce nedojde k úplnému ústupu příznaků nebo se příznaky objeví znovu, lze po dalších 6 hodinách podat třetí injekci přípravku Firazyr. V průběhu 24 hodin by neměly být podány více než 3 injekce přípravku Firazyr.V rámci klinických studií nebylo podáváno více než 8 injekcí přípravku Firazyr měsíčně. Pediatrická populace: Doporučená dávka přípravku Firazyr na základě tělesné hmotnosti u dětí a dospívajících (ve věku 2 až 17 let) je pro tělesnou hmotnost 12 kg až 25 kg 10 mg (1,0 ml), 26 kg až 40 kg 15 mg (1,5 ml), 41 kg až 50 kg, 20 mg (2,0 ml), 51 kg až 65 kg 25 mg (2,5 ml), > 65 kg 30 mg (3,0 ml). V klinické studii nebyla podána více než 1 injekce přípravku Firazyr na jednu ataku HAE. Starší osoby: Zkušenosti s podáváním přípravku u pacientů ve věku nad 65 let jsou omezené. U starších osob byla prokázána zvýšená systémová expozice ikatibantu. Není známo, zda je tato skutečnost významná ve vztahu k bezpečnosti přípravku Firazyr. Způsob podání: Firazyr je určen k subkutánnímu podání, nejlépe do břišní oblasti. Injekční roztok přípravku Firazyr by měl být injikován pomalu v důsledku objemu, který se podává. Každá stříkačka přípravku Firazyr je určena pouze pro jednorázové použití. O zahájení podávání přípravku Firazyr ošetřující osobou nebo samotným pacientem by měl rozhodnout pouze lékař se zkušenostmi v diagnostice a léčbě dědičného angioedému. Dospělí: Firazyr může být podáván samotným pacientem nebo ošetřující osobou pouze po proškolení v technice subkutánní injekce provedeném zdravotnickým pracovníkem. Děti a dospívající ve věku 2 – 17 let: Firazyr by měla podávat ošetřující osoba pouze po proškolení v technice subkutánní injekce provedeném zdravotnickým pracovníkem.
Kontraindikace: Hypersenzitivita na léčivou látku nebo na kteroukoli pomocnou látku tohoto přípravku.
Zvláštní upozornění: Laryngeální ataky: Pacienty s laryngeálními atakami je třeba po podání injekce pečlivě sledovat ve vhodném zdravotnickém zařízení, dokud lékař nerozhodne, že pacienta lze bez rizika propustit. Ischemická choroba srdeční: V případě ischémie může antagonismus bradykininových receptorů II. typu teoreticky způsobit zhoršení srdeční funkce a snížení průtoku krve koronárními cévami. Při podávání přípravku Firazyr pacientům s akutní ischemickou chorobou srdeční nebo nestabilní anginou pectoris je proto zapotřebí opatrnosti. Mozková příhoda: Ačkoli existují důkazy, které podporují pozitivní vliv blokády B2 receptorů bezprostředně po vzniku mozkové příhody, teoreticky je možné, že by ikatibant mohl oslabit pozitivní neuroprotektivní účinek bradykininu v pozdní fázi. Proto je zapotřebí opatrnosti při podávání ikatibantu pacientům během několika týdnů po vzniku mozkové příhody. Samostatné podávání pacientem nebo ošetřující osobou: Pacientům, kteří Firazyr nikdy dříve nedostali, by měla být první dávka podána ve zdravotnickém zařízení nebo pod dohledem lékaře. Pokud po samostatném podání nebo podání ošetřující osobou nedojde k úplnému ústupu příznaků nebo se příznaky objeví znovu, doporučuje se, aby pacient nebo ošetřující osoba vyhledali lékařskou pomoc. Pro dospělé by se měly následné dávky, které jsou nutné pro stejnou ataku, podat ve zdravotnickém zařízení. Pacienti s laryngeální atakou mají vždy vyhledat lékařskou pomoc a být sledováni ve zdravotnickém zařízení, a to i poté, co si aplikovali injekci v domácím prostředí. Existují omezené zkušenosti s léčbou více než jedné ataky HAE s použitím přípravku Firazyr u pediatrické populace.
Interakce: Neočekávají se žádné farmakokinetické lékové interakce s postižením CYP450. Současné podávání přípravku Firazyr s inhibitory angiotenzin konvertujícího enzymu (ACE) nebylo zkoumáno. ACE inhibitory jsou kontraindikovány u pacientů s dědičným angioedémem vzhledem k možnému zvýšení hladiny bradykininu.
Účinky na schopnost řídit a obsluhovat stroje: Pacientům je třeba doporučit, aby neřídili a neobsluhovali stroje, jestliže se cítí unaveni nebo mají-li závratě v důsledku ataky dědičného angioedému nebo po užití přípravku Firazyr.
Nežádoucí účinky: V klinických studiích použitých pro registraci bylo celkem 999 atak dědičného angioedému léčeno 30 mg Firazyru podanými subkutánně zdravotnickým pracovníkem. Téměř u všech jedinců, kteří byli v rámci klinických studií léčeni podkožně podávaným ikatibantem, se vyskytly reakce v místě podání injekce (charakterizované podrážděním kůže, otokem, bolestí, svěděním, erytémem, pocitem pálení). Tyto reakce byly zpravidla mírné až středně závažné, přechodné a vymizely bez nutnosti další intervence. Velmi časté (≥1/10): Reakce v místě injekce. Časté (≥1/100 až <1/10): Závrať, bolest hlavy, nauzea, vyrážka, erytém, pruritus, pyrexie, zvýšení transamináz. Není známo: Kopřivka. Pediatrická populace: Celkem 32 pediatrických pacientů s HAE bylo během klinických studií vystaveno léčbě ikatibantem. Přípravek Firazyr byl podán podkožní injekcí v dávce 0,4 mg/kg na základě tělesné hmotnosti do maximální dávky 30 mg.Většina pacientů, kteří byli léčeni podkožním ikatibantem měli reakci v místě injekce, jako je erytém, otok, pocit pálení, bolesti kůže a svědění nebo pruritus. Tyto reakce byly mírné až střední závažnosti a odpovídaly reakcím, které byly hlášené u dospělých. Dva pediatričtí pacienti měli reakci v místě injekce, která byla hodnocena jako závažná a zcela ustoupila během 6 hodin. Tyto reakce zahrnovaly erytém, otok, pálení a pocit tepla. Další informace o nežádoucích účincích jsou uvedeny v úplném znění Souhrnu údajů o přípravku.
Uchovávání: Uchovávejte při teplotě do 25 ○C. Chraňte před mrazem.
Obsah balení: Jedna předplněná injekční stříkačka s jednou jehlou nebo vícečetné balení obsahující tři předplněné injekční stříkačky se třemi jehlami. Na trhu nemusí být všechny velikosti balení.
Držitel rozhodnutí o registraci: Takeda Pharmaceuticals International AG Ireland Branch, Block 3 Miesian Plaza, 50 – 58 Baggot Street Lower, Dublin 2, D02 Y754, Irsko
Registrační čísla: EU/1/08/461/001, EU/1/08/461/002
Poslední revize SPC: 10/2021
Výdej léčivého přípravku je vázán na lékařský předpis.
Léčivý přípravek je hrazen z prostředků veřejného zdravotního pojištění.
Úplné znění SPC naleznete na www.sukl.cz.
Žádáme zdravotnické pracovníky, aby hlásili podezření na nežádoucí účinky na SÚKL nebo společnosti Takeda emailem na AE.CZE@takeda.com. Podezření na nežádoucí účinky hlaste také podle národních legislativních požadavků.
Endogenní parathormon (PTH) je vylučován příštítnými tělísky jako polypeptid z 84 aminokyselin. PTH působí prostřednictvím buněčných povrchových receptorů na parathormon, které jsou přítomné v kosti, ledvinách a nervové tkáni.
Parathormon má celou řadu zásadních fyziologických funkcí, které zahrnují jeho ústřední úlohu při regulaci hladiny kalcia a fosfátů v séru v pevně daných hladinách, regulaci vylučování kalcia a fosfátu ledvinami, aktivaci vitaminu D a udržování normálního kostního obratu.
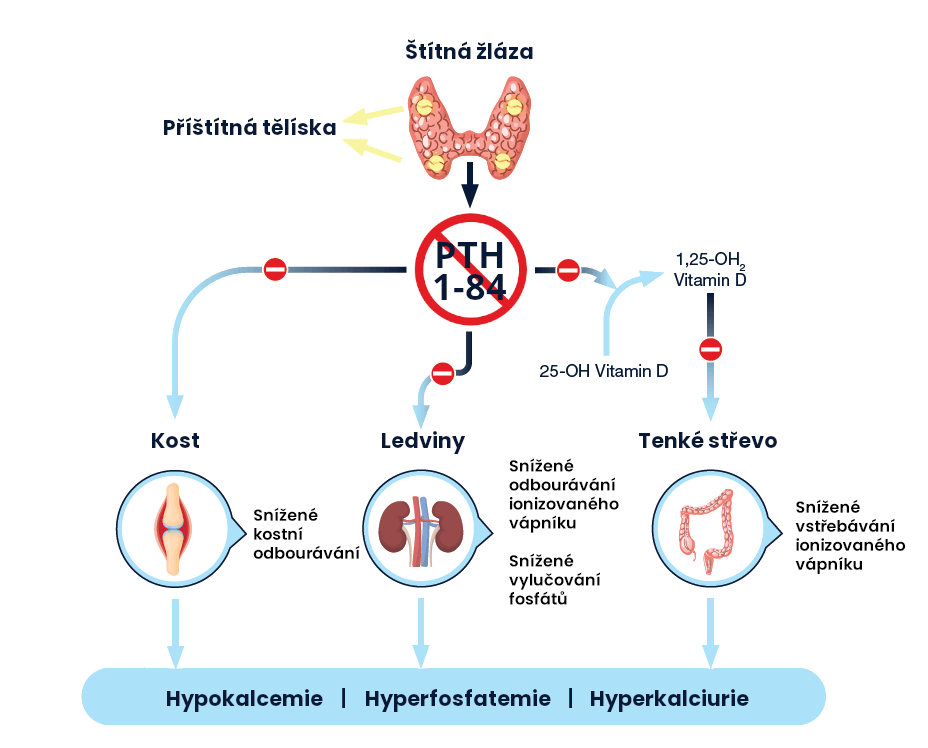
Natpar je produkovaný E. coli pomocí rekombinantní DNA technologie a je identický s 84 aminokyselinovou sekvencí endogenního lidského parathormonu. Natpar má stejnou primární aminokyselinovou sekvenci jako endogenní parathormon a je možné předpokládat, že má stejné fyziologické účinky.

PTH (1-84) je hlavním regulátorem homeostázy kalcia v plazmě. V ledvinách zvyšuje PTH (1-84) renální tubulární reabsorpci kalcia a podporuje vylučování fosfátů. Celkový účinek PTH je zvýšení koncentrace kalcia v séru, snížení vylučování kalcia močí a nižší koncentrace fosfátů v séru.
Přípravek Natpar je indikován jako přídatná léčba dospělých pacientů s chronickou hypoparathyreózou, kteří nejsou dostatečně kontrolováni samostatnou standardní léčbou.1
Standardní léčba chronické hypoparatyreózy spočívá především v denním užívání různých dávek doplňků vápníku a aktivního vitaminu D. To může udržovat hladinu vápníku na téměř normální úrovni, ale neřeší to základní nedostatek parathormonu, což znamená, že minerální homeostáza zůstává narušena a pacienti nadále trpí projevy nemoci. Navíc vysoké dávky vápníku a aktivního vitaminu D mohou vést k hyperkalciurii a usazeninám vápníku v ledvinách 2-4
Cílem léčby přípravkem Natpar je dosažení kontroly kalcémie, a tak snížení příznaků hypoparathyreózy. Jedná se o jediný léčivý přípravek, který může pomoci dané skupině pacientů, která je definovaná Českou endokrinologickou společností.1
DEFINICE PACIENTA S CHRONICKOU HYPOPARATYREÓZOU,
který není dostatečně kontrolován samostatnou standardní léčbou a u kterého by měla být zvážena substituční léčba rhPTH (1–84).1,3
Dosažení jednoho či více následujících kritérií Vám může pomoci identikovat nedostatečně kontrolovaného pacienta.1-4
SLEDOVANÁ KRITÉRIA
Současná léčba1-4

Denní perorální dávka převyšuje:
· 2,5 g kalcia NEBO · 1,5 µg kalcitriolu nebo 3 µg alfakalcidolu
Biochemické parametry1-4

· Nemožnost udržet optimální hladinu sérového kalcia v dolním pásmu referenčního rozmezí případně mírně pod dolní referenční mez, bez dokumentovaných epizod hyperkalcemie

· Opakující se hyperkalciurie > 7,5 mmol/24 h u mužů a > 6,25 mmol/24 h u žen
· Snížení glomerulární filtrace (GF) < 1,0 ml/s/1,73 m² (< 60 ml/min/1,73 m²)

· Hyperfosfatemie A/NEBO kalcio-fosfátový produkt > 4,4 mmol2/l2
Život ohrožující symptomy spojené s hypoparatyreózou1

Broncho/laryngospasmus
Arytmie
Generalizované křeče
Jiné parametry1-4

· Komorbidity jako je nefrolitiáza nebo o nefrokalcinóza dle zobrazovacích metod
· Prokazatelná klinicky závažná malabsorpce GIT
Výrazně omezená kvalita života z důvodu přítomnosti závažných symptomů hypokalcemie1-4

Reference: 1. Vnitr Lek 2021, 67(Suppl.A) 2. Khan AA, et al. Eur J Endocrinol. 2019; 180(3):P1–22. 3. Bilezikian JP, et al. J Clin Endocrinol Metab. 2016(10);101:2313–24.
4. Brandi ML, et al. J Clin Endocrinol Metab. 2016;101(6):2273–83. 5. Tabacco G, Tay Y-KD, Cusano NE, et al. Quality of life in hypoparathyroidism improves with rhPTH(1-84) throughout 8 years of therapy. J Clin Endocrinol Metab. 2019;104:2748-2756.
Před zahájením léčby přípravkem Natpar
- Natpar je vhodný pro podávání samotným pacientem. Každá dávka se musí podávat jako subkutánní injekce jednou denně, střídavě do každého stehna.1
- Pacienti musí být proškoleni předepisujícím lékařem nebo zdravotnickým odborným personálem o správném způsobu aplikace.1
4 různá dávkování – stejná cena

Léčba přípravkem Natpar vyžaduje nastavení a zahájení léčby, úpravu dávky a sledování
Před zahájením léčby přípravkem Natpar se seznamte s úplným souhrnem údajů o přípravku.

NASTAVENÍ A ZAHÁJENÍ LÉČBY PŘÍPRAVKEM NATPAR1
PŘED zahájením léčby
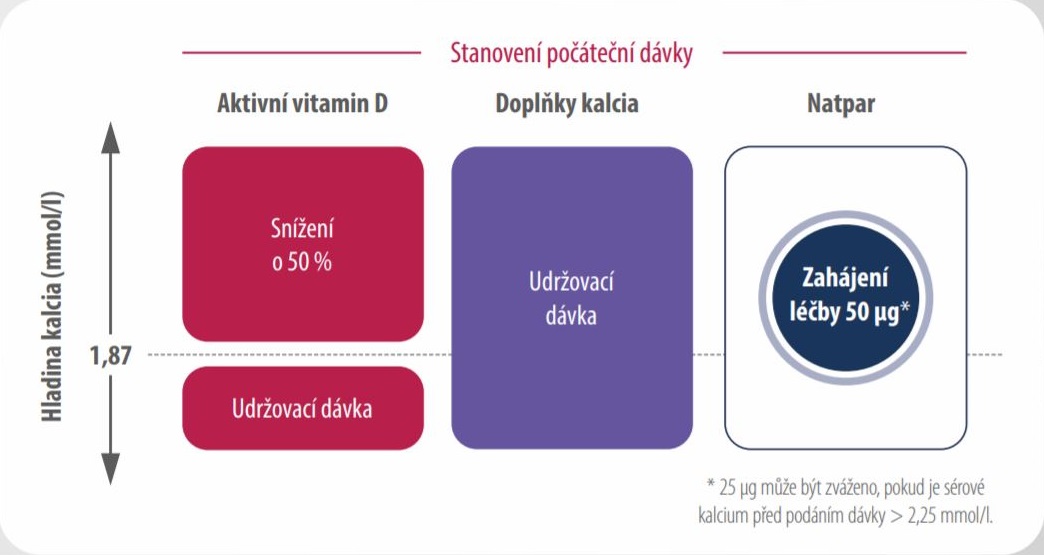
- Ujistěte se o dostatečném stavu zásobení 25-OH vitaminem D
- Ověřte, že sérové kalcium a magnesium jsou v referenčním rozmezí
- Dle hodnoty sérového kalcia se rozhodněte o počáteční dávce přípravku Natpar a aktivního vitaminu D
ZAHÁJENÍ léčby
Počáteční dávka přípravku Natpar: léčba se zahajuje dávkou 50 µg jednou denně subkutánní injekcí do jednoho ze stehen. Pokud je před podáním dávky sérové kalcium >2,25 mmol/l, je možné zvážit podání počáteční dávky 25 μg.- hladina sérového kalcia před dávkou je v rozmezí 2,0–2,25 mmol/l
- snížení nebo přerušení suplementace aktivním vitaminem D
- dostatečná suplementace kalcia
Měření požadované před zahájením léčby přípravkem Natpar
- Aktivní vitamin D: Pokud je sérové kalcium před podáním dávky > 1,87 mmol/l a pacient užívá aktivní vitamin D, snižte jeho dávku o 50 %.
- Kalcium: U pacientů, kteří užívají doplňky kalcia, udržujte jejich dávku.

ÚPRAVA DÁVKY PŘÍPRAVKU NATPAR V PRŮBĚHU LÉČBY1
I. Úprava dávky PO zahájení léčby
Kontrolní odběry v rozmezí 2–5 dnů po první aplikaci přípravku Natpar
- Změřte sérové kalcium před podáním pravidelné dávky (24 hodin po poslední aplikaci), a pokud je hladina sérového kalcia < 1,87 mmol/l nebo > 2,55 mmol/l zopakujte měření následující den.
- Upravte dávku aktivního vitaminu D nebo doplňku kalcia (nebo obou) na základě sérové hladiny kalcia a Vašeho klinického hodnocení (tzn. příznaky hypokalcemie nebo hyperkalcemie).
II. Úprava dávky V PRŮBĚHU léčby (titrace)
Kontrolní odběry po 14–28 dnech od první aplikace přípravku Natpar
Dávka přípravku Natpar se může zvýšit o 25 µg každé 2–4 týdny, do dosažení maximální denní dávky 100 µg. Snížení dávky na minimálních 25 µg může proběhnout kdykoli. Pro optimalizaci léčby se doporučuje změřit hladinu sérového kalcia korigovanou na albumin 8–12 hodin po podání přípravku Natpar (maximální hodnota). Před plánovaným zvýšením dávky přípravku Natpar je doporučeno opakované měření hladiny sérového kalcia korigovaného na albumin před a po aplikaci přípravku Natpar.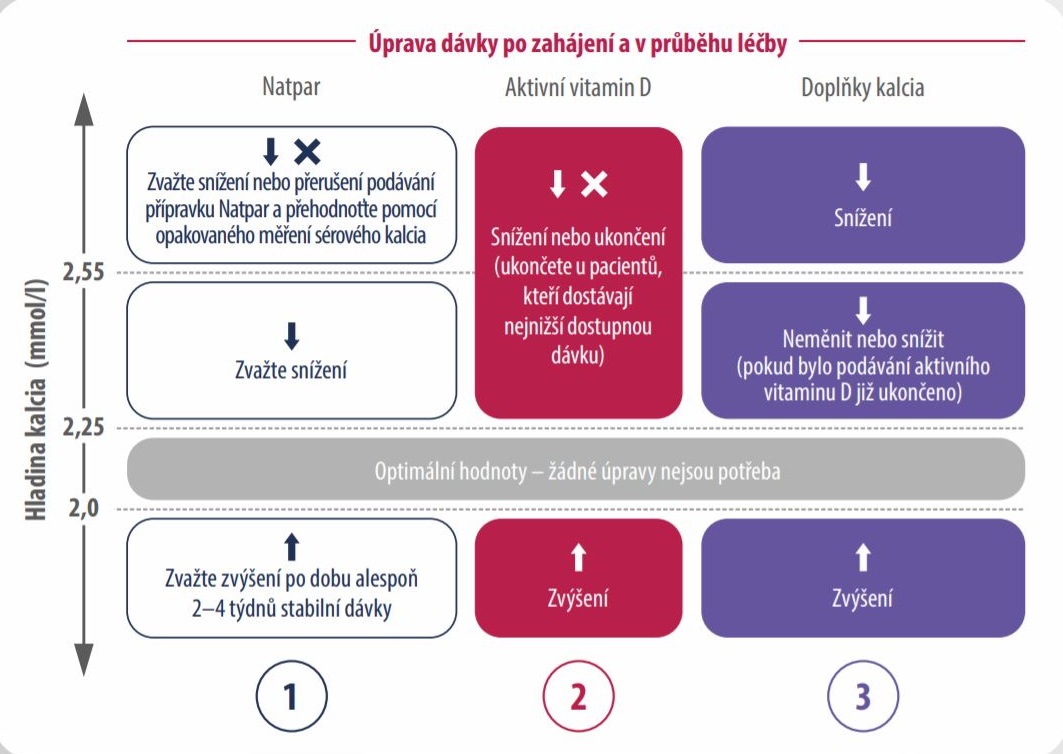
Data z klinických studií ukazují, že u více jak poloviny pacientů léčených přípravkem Natpar je dávka upravena na maximální možnou dávku 100 µg denně.5
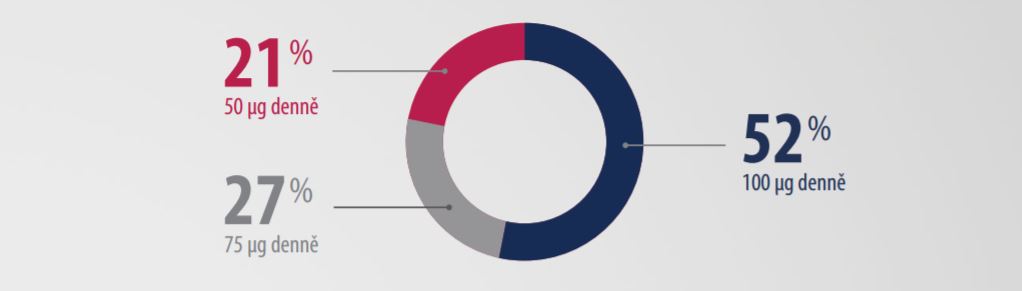
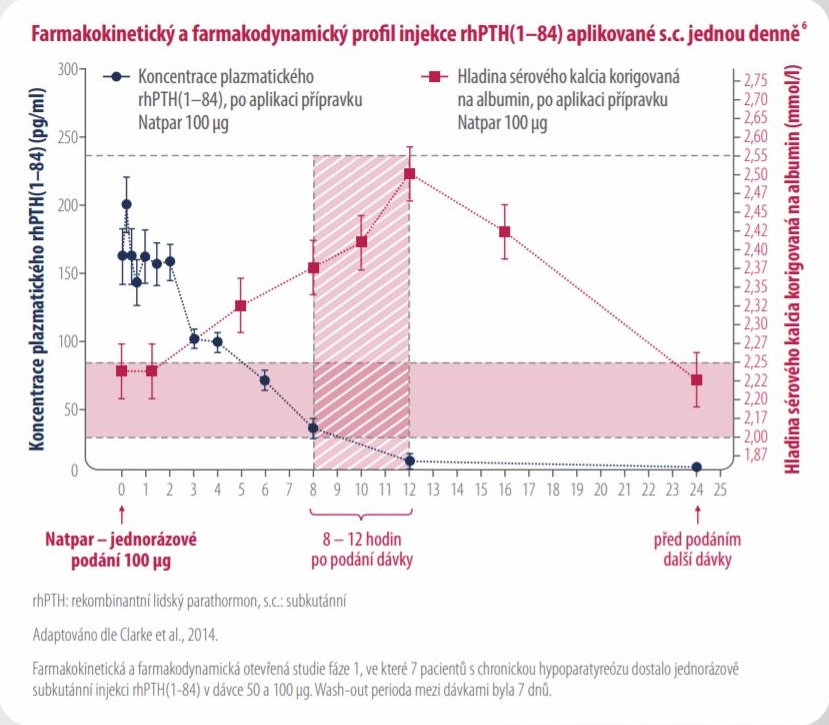
Natpar podávaný jednou denně zajišťuje stabilní hladinu kalcia po dobu 24 hodin1,6
 Dočasné nebo trvalé přerušení léčby přípravkem Natpar musí být doprovázeno monitorováním hladiny kalcia v séru
a zvýšením exogenního kalcia a/nebo podle potřeby zdrojů aktivního vitaminu D.
Dočasné nebo trvalé přerušení léčby přípravkem Natpar musí být doprovázeno monitorováním hladiny kalcia v séru
a zvýšením exogenního kalcia a/nebo podle potřeby zdrojů aktivního vitaminu D.

DLOUHODOBÉ SLEDOVÁNÍ PACIENTA
Doporučené pravidelné sledování:7

U pacientů s chronickou hypoparatyreózou se mohou častěji vyskytnout tyto komorbidity:

Evropská endokrinologická společnost doporučuje individualizovanou léčbu a sledování7
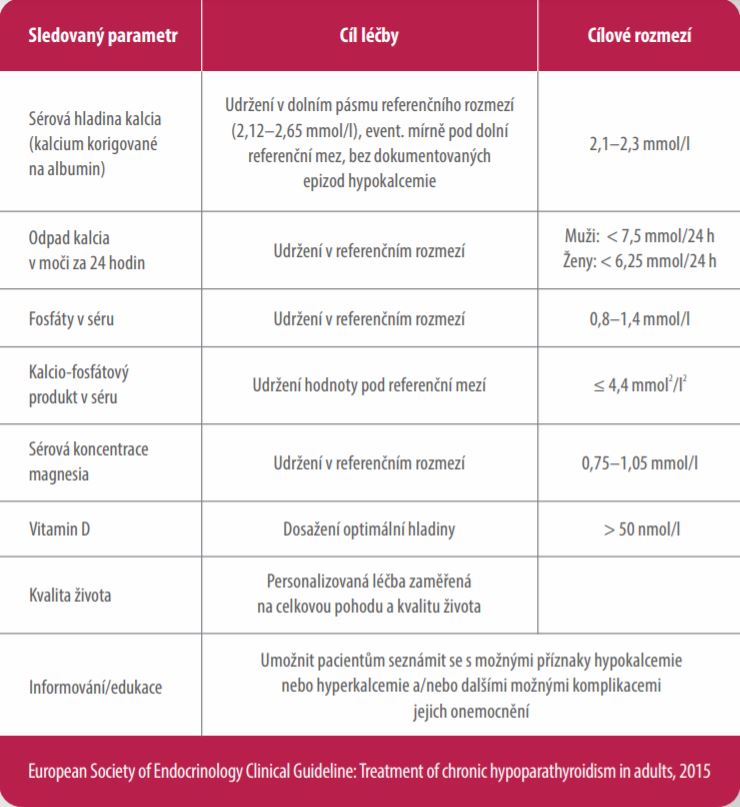
Cílem léčby přípravkem Natpar je dosažení kontroly kalcemie, a tak snížení příznaků hypoparatyreózy společně s optimalizací parametrů kalcio-fosfátového metabolismu1,7
Cíle léčby u pacientů s chronickou hypoparatyreózou7,10
HYPOPARA PROGRAM
Zahájení a průběh léčby LP Natpar
Vhodný pacient

Pacient, který není
dostatečně kontrolován
samostatnou standardní
léčbou dle kritérií
schválených
Českou endokrinologickou
společností.


ZDRAVOTNICKÝ
PERSONÁL
Proškolení
zdravotnického
personálu
ke správnému
používání
pera Natpar
zástupcem
společnosti
Takeda.

Schválení léčby

Obdržení informace
o schválení léčby.
Volat pacientovi

Telefonická domluva
termínu prvního podání
LP Natpar a proškolení
správného způsobu aplikace.
Připomenout registraci
do HYPOPARA programu.
Kontrolní odběry

Před zahájením
léčby přípravkem
Natpar.
Pero Natpar

Vyzvednutí 2 ks v lékárně.
1 pero Natpar je náhradní.
1. den léčby

Edukace a aplikace
Proškolení správného
způsobu aplikace
+ první aplikace.
2–5 dnů od zahájení léčby

Kontrolní odběry

Případná úprava
dávky aktivního
vitaminu D nebo kalcia
(nebo obou).
14-28 dnů po zahájení léčby
Kontrolní odběry
Dávku přípravku Natpar
lze zvýšit o 25 µg
každé 2–4 týdny.
Snížení může
proběhnout kdykoliv.
Každých 3–6 měsíců

Kontrolní odběry

Sledování biochemických
parametrů (hladina
sérového kalcia, magnesia,
fosfátů a kreatininu, eGFR),
sledování příznaků
hypokalcemie
nebo hyperkalcemie.
1× ročně
Kontrolní odběry
Sledování
Odpadu kalcia v moči
za 24 hodin
a sledování projevů
a příznaků komorbidit.

PACIENT

Info letáček
Pacient obdrží v ordinaci lékaře
informace k účasti
v HYPOPARA programu.

Registrace do programu
Registrace do HYPOPARA
programu dle instrukcí
v letáčku. Registraci je nutné
udělat v okamžiku,
kdy bude znám termín prvního
podání LP Natpar u ošetřujícího
endokrinologa včetně proškolení.

Edukační balíček
Na základě registrace
do HYPOPARA programu
odeslání edukačního
balíčku na adresu pacienta
(do dvou pracovních dnů).
Obdržení edukačního balíčku
s doporučením
pečlivého prostudování
návodů k aplikaci.
2. den léčby
1. telefonická konzultace
Telefonická konzultace
v rámci HYPOPARA programu
s cílem pomoci s první
samostatnou aplikací
v domácím prostředí.
13. den od zahájení léčby
2. telefonická konzultace
Telefonická konzultace
v rámci HYPOPARA programu
s cílem připomenout postup
při smíchání nové náplně
následující den.
3 měsíce od zahájení léčby
3. telefonická konzultace
Závěrečná telefonická
konzultace v rámci
HYPOPARA programu.
Po roce léčby

Dotazník spokojenosti
Elektronický dotazník
spojenosti v rámci
HYPOPARA programu.

Exspirace pera
Automatické
upozornění na konec
exspirace pera.
HYPOPARA PROGRAM
Zahájení a průběh léčby LP Natpar
Vhodný pacient
Pacient, který není
dostatečně kontrolován
samostatnou standardní
léčbou dle kritérií
schválených
Českou endokrinologickou
společností.
ZDRAVOTNICKÝ
PERSONÁL
Proškolení
zdravotnického
personálu
ke správnému
používání
pera Natpar
zástupcem
společnosti
Takeda.
Schválení léčby
Obdržení informace
o schválení léčby.
Volat pacientovi
Telefonická domluva
termínu prvního podání
LP Natpar a proškolení
správného způsobu aplikace.
Připomenout registraci
do HYPOPARA programu.
Kontrolní odběry
Před zahájením
léčby přípravkem
Natpar.
Pero Natpar
Vyzvednutí 2 ks v lékárně.
1 pero Natpar je náhradní.
1. den léčby
Edukace a aplikace
Proškolení správného
způsobu aplikace
+ první aplikace.
2–5 dnů od zahájení léčby
Kontrolní odběry
Případná úprava
dávky aktivního
vitaminu D nebo kalcia
(nebo obou).
14-28 dnů po zahájení léčby
Kontrolní odběry
Dávku přípravku Natpar
lze zvýšit o 25 µg
každé 2–4 týdny.
Snížení může
proběhnout kdykoliv.
Každých 3–6 měsíců
Kontrolní odběry
Sledování biochemických
parametrů (hladina
sérového kalcia, magnesia,
fosfátů a kreatininu, eGFR),
sledování příznaků
hypokalcemie
nebo hyperkalcemie.
1× ročně
Kontrolní odběry
Sledování
Odpadu kalcia v moči
za 24 hodin
a sledování projevů
a příznaků komorbidit.
PACIENT
Info letáček
Pacient obdrží v ordinaci lékaře
informace k účasti
v HYPOPARA programu.
Registrace do programu
Registrace do HYPOPARA programu dle instrukcí v letáčku. Registraci je nutné udělat v okamžiku, kdy bude znám termín prvního podání LP Natpar u ošetřujícího endokrinologa včetně proškolení.
Edukační balíček
Na základě registrace
do HYPOPARA programu
odeslání edukačního
balíčku na adresu pacienta
(do dvou pracovních dnů).
Obdržení edukačního balíčku
s doporučením
pečlivého prostudování
návodů k aplikaci.
2. den léčby
1. telefonická konzultace
Telefonická konzultace v rámci HYPOPARA programu s cílem pomoci s první samostatnou
aplikací v domácím
prostředí.
13. den od zahájení léčby
2. telefonická konzultace
Telefonická konzultace
v rámci HYPOPARA programu
s cílem připomenout postup
při smíchání nové náplně
následující den.
3 měsíce od zahájení léčby
3. telefonická konzultace
Závěrečná telefonická
konzultace v rámci
HYPOPARA programu.
Po roce léčby
Dotazník spokojenosti
Elektronický dotazník
spojenosti v rámci
HYPOPARA programu.
Exspirace pera
Automatické
upozornění na konec
exspirace pera.
Pro zobrazení časové osy otočte vaše mobilní zařízení
Reference: 1. SPC přípravku Natpar, poslední revize textu duben 2020. 2. Bollerslev J, Rejnmark L, et al. European Society of Endocrinology Clinical Guideline: Treatment of chronic hypoparathyroidism in adults. Eur J Endocrinol 2015;173(2):G1-20.
C-APROM/CZ/NATP/0013 • Datum přípravy: únor 2021
Mohlo by Vás zajímat




NATPAR
Zkrácené informace o léčivém přípravku
▼ Tento léčivý přípravek podléhá dalšímu sledování. To umožní rychlé získání nových informací o bezpečnosti. Žádáme zdravotnické pracovníky, aby hlásili jakákoli podezření na nežádoucí účinky. Podrobnosti o hlášení nežádoucích účinků viz bod 4.8 SPC.
Název přípravku: Natpar 25 mikrogramů/dávka prášek a rozpouštědlo pro injekční roztok; Natpar 50 mikrogramů/dávka prášek a rozpouštědlo pro injekční roztok; Natpar 75 mikrogramů/dávka prášek a rozpouštědlo pro injekční roztok; Natpar 100 mikrogramů/dávka prášek a rozpouštědlo pro injekční Složení: Jedna dávka obsahuje po rekonstituci 25/50/75/100 mikrogramů hormonum parathyroidum (rDNA)* v 71,4 mikrolitrech roztoku. Jedna zásobní vložka obsahuje 350/700/1050/1400 mikrogramů hormonum parathyroidum (rDNA). *Parathormon (rDNA) produkovaný E. coli pomocí rekombinantní DNA technologie je identický se sekvencí 84 aminokyselin endogenního lidského parathormonu. Indikace: Přípravek Natpar je indikován jako přídatná léčba dospělých pacientů s chronickou hypoparathyreózou, kteří nejsou dostatečně kontrolováni samostatnou standardní léčbou. Dávkování a způsob podání: Zahajte léčbu 50 mikrogramy jednou denně subkutánní injekcí do stehna (střídejte stehna každý den). Pokud je před podáním dávky sérové kalcium > 2,25 mmol/l, je možné zvážit podání počáteční dávky 25 mikrogramů. U pacientů užívajících aktivní vitamin D snižte jeho dávku o 50 %, pokud je sérové kalcium před podáním dávky nad 1,87 mmol/l. Udržujte doplňkovou dávku kalcia. Změřte sérovou koncentraci kalcia během 2 až 5 dnů před podáním dávky. Pokud je sérová hladina kalcia před dávkou pod 1,87 mmol/l nebo nad 2,55 mmol/l, mělo by se měření opakovat následující den. Upravte dávku aktivního vitamínu D nebo doplňku kalcia nebo obou na základě sérové hladiny kalcia a klinického hodnocení. Úpravy se mají opakovat, dokud nebudou cílové hladiny sérového kalcia před dávkou v rozsahu 2,0 – 2,25 mmol/l, aktivní vitamín D bude ukončen a doplněk kalcia je dostatečný pro splnění denních požadavků. Během titrace musí být monitorována koncentrace sérového kalcia. Dávka přípravku Natpar se může zvýšit o 25 mikrogramů přibližně každé 2 až 4 týdny, do maximální denní dávky 100 mikrogramů. K titraci dávky dolů na minimum 25 mikrogramů může dojít kdykoliv. Pokyny k úpravě dávky po úvodním období – viz SPC. Přípravek Natpar je vhodný pro podávání samotným pacientem jako subkutánní injekce jednou denně střídavě do každého stehna. Kontraindikace: Hypersenzitivita na léčivou látku nebo na kteroukoli pomocnou látku. U pacientů, kteří podstupují nebo dříve podstupovali radioterapii skeletu, s malignitami skeletu nebo kostními metastázami, kteří mají zvýšené výchozí riziko vzniku osteosarkomu (pacienti s Pagetovou chorobou kostí nebo dědičnými poruchami), s nevysvětleným zvýšením kostní alkalické fosfatázy a s pseudohypoparathyreózou. Zvláštní upozornění a opatření pro použití: Monitorování pacientů během léčby: Sérové hladiny kalcia před dávkou a v některých případech po dávce musí být během léčby přípravkem Natpar sledovány. Hyperkalcemie: často se objevovala během titračního období. V případě závažné hyperkalcemie (> 3,0 mmol/l nebo nad horní hranici normy s příznaky), zvažte hydrataci a dočasné zastavení podávání přípravku Natpar, kalcia a aktivního vitamínu D, dokud se sérové kalcium nevrátí do normálního rozsahu. Hypokalcemie: Riziko závažné hypokalcemie je nejvyšší po odložení podání, vynechání nebo náhlém vysazení přípravku Natpar, může se ale vyskytnout kdykoli. Souběžné použití se srdečními glykosidy: Hyperkalcemie z jakékoliv příčiny může predisponovat k toxicitě digitalis. Závažná porucha funkce ledvin nebo jater: Používat s opatrností u pacientů se závažnou poruchou ledvin nebo jater. Použití u mladých dospělých: Používat s opatrností u mladých dospělých pacientů s otevřenými epifýzami, protože tito pacienti mohou být vystaveni zvýšenému riziku vzniku osteosarkomu. Použití u starších pacientů: Klinické studie nezahrnovaly dostatečný počet subjektů ve věku ≥ 65 let pro stanovení, zda je odpověď u těchto subjektů odlišná od mladších subjektů. Tachyfylaxe: Účinek přípravku Natpar se může po čase u některých pacientů snížit. Urolitiáza: U pacientů s aktivní nebo nedávnou urolitiázou je nutno používat přípravek Natpar s opatrností vzhledem k jeho potenciálu tento stav zhoršovat. Hypersenzitivita: Po uvedení na trh byly hlášeny hypersenzitivní reakce, které mohou zahrnovat anafylaxi, dyspnoi, angioedém, kopřivku, vyrážku atd. Interakce: Kombinované použití přípravku Natpar a srdečních glykosidů může pacienty predisponovat k toxicitě digitalisu, pokud vznikne hyperkalcémie. Současné používání přípravku Natpar s bisfosfonáty se nedoporučuje. Fertilita, těhotenství a kojení: Údaje o podávání přípravku Natpar těhotným ženám nejsou k dispozici. Studie reprodukční toxicity na zvířatech nenaznačují přímé nebo nepřímé škodlivé účinky. Není známo, zda se přípravek Natpar vylučuje do lidského mateřského mléka. Dostupné farmakologické údaje u zvířat prokázaly vylučování přípravku Natpar do mléka. Neexistují žádné údaje u člověka o vlivu přípravku Natpar na plodnost. Studie na zvířatech nenaznačují žádnou poruchu plodnosti. Nežádoucí účinky: Nejčastější nežádoucí reakce byly hyperkalcemie, hypokalcemie a jejich související klinické projevy, zahrnující bolesti hlavy, průjem, zvracení, parestezie, hypestezie a hyperkalciurie. Ostatní nežádoucí účinky – viz SPC. Předávkování: Může způsobit hyperkalcémii. Těžká hyperkalcémie může být život ohrožující stav vyžadující neodkladnou lékařskou péči a pečlivé monitorování. Podmínky uchovávání: Uchovávejte v chladničce (2 °C – 8 °C). Chraňte před mrazem. Uchovávejte zásobní vložky v držáku v krabičce, aby byly chráněny před světlem. Podmínky uchovávání po rekonstituci – viz SPC. Držitel rozhodnutí o registraci: Shire Pharmaceuticals Ireland Limited, Block 2 & 3 Miesian Plaza, 50 – 58 Baggot Street Lower, Dublin 2, Irsko. Registrační čísla: EU/1/15/1078/001-004. Poslední revize SPC: 04/2020.
Výdej léčivého přípravku je vázán na lékařský předpis. Léčivý přípravek není hrazen z prostředků veřejného zdravotního pojištění. Před předepsáním se seznamte s úplným zněním Souhrnu údajů o přípravku.
Žádáme zdravotnické pracovníky, aby hlásili podezření na nežádoucí účinky na SÚKL nebo společnosti Takeda emailem na AE.CZE@takeda.com. Podezření na nežádoucí účinky hlaste také podle národních legislativních požadavků.
▼ Tento léčivý přípravek podléhá dalšímu sledování. To umožní rychlé získání nových informací o bezpečnosti. Žádáme zdravotnické pracovníky, aby hlásili jakákoli podezření na nežádoucí účinky. Podrobnosti o hlášení nežádoucích účinků viz bod 4.8 SPC.
Název přípravku: Natpar 25 mikrogramů/dávka prášek a rozpouštědlo pro injekční roztok; Natpar 50 mikrogramů/dávka prášek a rozpouštědlo pro injekční roztok; Natpar 75 mikrogramů/dávka prášek a rozpouštědlo pro injekční roztok; Natpar 100 mikrogramů/dávka prášek a rozpouštědlo pro injekční Složení: Jedna dávka obsahuje po rekonstituci 25/50/75/100 mikrogramů hormonum parathyroidum (rDNA)* v 71,4 mikrolitrech roztoku. Jedna zásobní vložka obsahuje 350/700/1050/1400 mikrogramů hormonum parathyroidum (rDNA). *Parathormon (rDNA) produkovaný E. coli pomocí rekombinantní DNA technologie je identický se sekvencí 84 aminokyselin endogenního lidského parathormonu. Indikace: Přípravek Natpar je indikován jako přídatná léčba dospělých pacientů s chronickou hypoparathyreózou, kteří nejsou dostatečně kontrolováni samostatnou standardní léčbou. Dávkování a způsob podání: Zahajte léčbu 50 mikrogramy jednou denně subkutánní injekcí do stehna (střídejte stehna každý den). Pokud je před podáním dávky sérové kalcium > 2,25 mmol/l, je možné zvážit podání počáteční dávky 25 mikrogramů. U pacientů užívajících aktivní vitamin D snižte jeho dávku o 50 %, pokud je sérové kalcium před podáním dávky nad 1,87 mmol/l. Udržujte doplňkovou dávku kalcia. Změřte sérovou koncentraci kalcia během 2 až 5 dnů před podáním dávky. Pokud je sérová hladina kalcia před dávkou pod 1,87 mmol/l nebo nad 2,55 mmol/l, mělo by se měření opakovat následující den. Upravte dávku aktivního vitamínu D nebo doplňku kalcia nebo obou na základě sérové hladiny kalcia a klinického hodnocení. Úpravy se mají opakovat, dokud nebudou cílové hladiny sérového kalcia před dávkou v rozsahu 2,0 – 2,25 mmol/l, aktivní vitamín D bude ukončen a doplněk kalcia je dostatečný pro splnění denních požadavků. Během titrace musí být monitorována koncentrace sérového kalcia. Dávka přípravku Natpar se může zvýšit o 25 mikrogramů přibližně každé 2 až 4 týdny, do maximální denní dávky 100 mikrogramů. K titraci dávky dolů na minimum 25 mikrogramů může dojít kdykoliv. Pokyny k úpravě dávky po úvodním období – viz SPC. Přípravek Natpar je vhodný pro podávání samotným pacientem jako subkutánní injekce jednou denně střídavě do každého stehna. Kontraindikace: Hypersenzitivita na léčivou látku nebo na kteroukoli pomocnou látku. U pacientů, kteří podstupují nebo dříve podstupovali radioterapii skeletu, s malignitami skeletu nebo kostními metastázami, kteří mají zvýšené výchozí riziko vzniku osteosarkomu (pacienti s Pagetovou chorobou kostí nebo dědičnými poruchami), s nevysvětleným zvýšením kostní alkalické fosfatázy a s pseudohypoparathyreózou. Zvláštní upozornění a opatření pro použití: Monitorování pacientů během léčby: Sérové hladiny kalcia před dávkou a v některých případech po dávce musí být během léčby přípravkem Natpar sledovány. Hyperkalcemie: často se objevovala během titračního období. V případě závažné hyperkalcemie (> 3,0 mmol/l nebo nad horní hranici normy s příznaky), zvažte hydrataci a dočasné zastavení podávání přípravku Natpar, kalcia a aktivního vitamínu D, dokud se sérové kalcium nevrátí do normálního rozsahu. Hypokalcemie: Riziko závažné hypokalcemie je nejvyšší po odložení podání, vynechání nebo náhlém vysazení přípravku Natpar, může se ale vyskytnout kdykoli. Souběžné použití se srdečními glykosidy: Hyperkalcemie z jakékoliv příčiny může predisponovat k toxicitě digitalis. Závažná porucha funkce ledvin nebo jater: Používat s opatrností u pacientů se závažnou poruchou ledvin nebo jater. Použití u mladých dospělých: Používat s opatrností u mladých dospělých pacientů s otevřenými epifýzami, protože tito pacienti mohou být vystaveni zvýšenému riziku vzniku osteosarkomu. Použití u starších pacientů: Klinické studie nezahrnovaly dostatečný počet subjektů ve věku ≥ 65 let pro stanovení, zda je odpověď u těchto subjektů odlišná od mladších subjektů. Tachyfylaxe: Účinek přípravku Natpar se může po čase u některých pacientů snížit. Urolitiáza: U pacientů s aktivní nebo nedávnou urolitiázou je nutno používat přípravek Natpar s opatrností vzhledem k jeho potenciálu tento stav zhoršovat. Hypersenzitivita: Po uvedení na trh byly hlášeny hypersenzitivní reakce, které mohou zahrnovat anafylaxi, dyspnoi, angioedém, kopřivku, vyrážku atd. Interakce: Kombinované použití přípravku Natpar a srdečních glykosidů může pacienty predisponovat k toxicitě digitalisu, pokud vznikne hyperkalcémie. Současné používání přípravku Natpar s bisfosfonáty se nedoporučuje. Fertilita, těhotenství a kojení: Údaje o podávání přípravku Natpar těhotným ženám nejsou k dispozici. Studie reprodukční toxicity na zvířatech nenaznačují přímé nebo nepřímé škodlivé účinky. Není známo, zda se přípravek Natpar vylučuje do lidského mateřského mléka. Dostupné farmakologické údaje u zvířat prokázaly vylučování přípravku Natpar do mléka. Neexistují žádné údaje u člověka o vlivu přípravku Natpar na plodnost. Studie na zvířatech nenaznačují žádnou poruchu plodnosti. Nežádoucí účinky: Nejčastější nežádoucí reakce byly hyperkalcemie, hypokalcemie a jejich související klinické projevy, zahrnující bolesti hlavy, průjem, zvracení, parestezie, hypestezie a hyperkalciurie. Ostatní nežádoucí účinky – viz SPC. Předávkování: Může způsobit hyperkalcémii. Těžká hyperkalcémie může být život ohrožující stav vyžadující neodkladnou lékařskou péči a pečlivé monitorování. Podmínky uchovávání: Uchovávejte v chladničce (2 °C – 8 °C). Chraňte před mrazem. Uchovávejte zásobní vložky v držáku v krabičce, aby byly chráněny před světlem. Podmínky uchovávání po rekonstituci – viz SPC. Držitel rozhodnutí o registraci: Shire Pharmaceuticals Ireland Limited, Block 2 & 3 Miesian Plaza, 50 – 58 Baggot Street Lower, Dublin 2, Irsko. Registrační čísla: EU/1/15/1078/001-004. Poslední revize SPC: 04/2020.
Výdej léčivého přípravku je vázán na lékařský předpis. Léčivý přípravek není hrazen z prostředků veřejného zdravotního pojištění. Před předepsáním se seznamte s úplným zněním Souhrnu údajů o přípravku.
Žádáme zdravotnické pracovníky, aby hlásili podezření na nežádoucí účinky na SÚKL nebo společnosti Takeda emailem na AE.CZE@takeda.com. Podezření na nežádoucí účinky hlaste také podle národních legislativních požadavků.